HIF1α-NF-kB Pathway
调节HIF1α和NFkB在癌症治疗中的作用:这是控制恶性进展的合理方法吗?
Modulators of HIF1α and NFkB in cancer treatment: is it a
rational approach for controlling malignant progression?
连接缺氧、HIF1α激活、炎症和肿瘤恶化的示意图
NF-kB signaling pathway
什么是NF-kB?
NF-kB是活化的B细胞的核因子κ轻链增强剂的缩写。它不是单个蛋白质,而是一类小的可诱导的转录因子,在几乎所有哺乳动物细胞中都起着重要作用。它控制DNA转录,细胞因子产生,细胞存活和其他重要的细胞事件,尤其在调节对感染的免疫反应中起关键作用。
NF-kB分子通常是二聚体。 NF-kB的典型结构是P50-P65二聚体(NF-kB1 / RelA)。
DNA结合必须形成二聚体,两个NF-κB单体以二聚体的形式结合到DNA。二聚体的N末端区域负责特定的DNA接触。
C端区域通常是高度保守的,它们负责二聚化和非特异性DNA磷酸接触。整个NF-kB分子就像DNA链上的钳子钳一样,起着转录因子的作用(图2)。
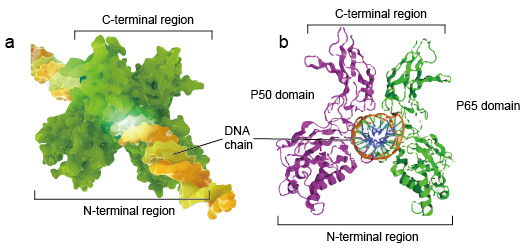
Figure 2. The structure of NF-kB protein dimer binding with DNA chain.
NF-kB家族
因为NF-κB家族成员与逆转录病毒癌蛋白v-Rel具有结构同源性。所以他们也叫NF-kB /
Rel蛋白。哺乳动物中的NF-kB转录因子家族由5种蛋白质组成:p65 / RelA,RelB,c-Rel,p105 / p50(NF-kB1)和p100 /
p52(NF-kB2)。这5种蛋白质相互结合,形成独特的转录活性同型/异二聚体复合物(图3)。它们都共享一个300
aa长度的共同保守Rel同源域(RHD)。这些RHD结构域具有多种功能,例如二聚化,DNA结合,与IkB的相互作用以及核易位。尽管NF-kB蛋白家族成员最多可以形成15个不同的二聚体,但其中许多尚未得到证实。
NF-kB二聚体最丰富的形式是p50 / p65异二聚体,已在几乎所有细胞类型中得到资助。由于仅P65 /
Rel,RelB和c-Rel具有羧基末端反式激活结构域(TAD),因此NF-kB蛋白家族可以进一步分为两组。
p50和p52分别通过处理前体分子p105和p100生成。因此,并非Rel二聚体的所有组合都具有转录活性。
.jpg)
Figure 3. The 5 protein and their homo/heterodimeric complexes of NF-kB family.
The NF-kB Signaling Pathway - Creative Diagnostics
https://www.creative-diagnostics.com/The-NF-kB-Signaling-Pathway.htm
NF-kB信号通路
NF-kB蛋白二聚体作为核转录因子,它们需要迁移到细胞核,与DNA结合才能发挥功能。在大多数处于静止状态的正常细胞中,NF-KB处于非活性状态并保留在细胞质中。它们与称为IK-B蛋白的特定抑制剂结合,该抑制剂可与NF-kB的Rel同源结构域(RHD)结合并干扰其核定位序列(NLS)的功能。这些抑制剂蛋白,包括IkBa,IkBb和IkBg,含有6-7个锚蛋白重复序列,介导与RHD的结合。这些重复序列也存在于NF-kB2
/ p100和NF-kB1 /
p105前体的C末端一半中,它们也起着IkB的作用并将其伴侣Rel蛋白保留在细胞质中。为了激活NF-kB分子,细胞首先需要从其抑制剂中分离出NF-kB蛋白。有两种主要的信号通路导致IK-B蛋白抑制剂从NF-kB二聚体解离,并使NF-kB二聚体从细胞质转移到细胞核中。
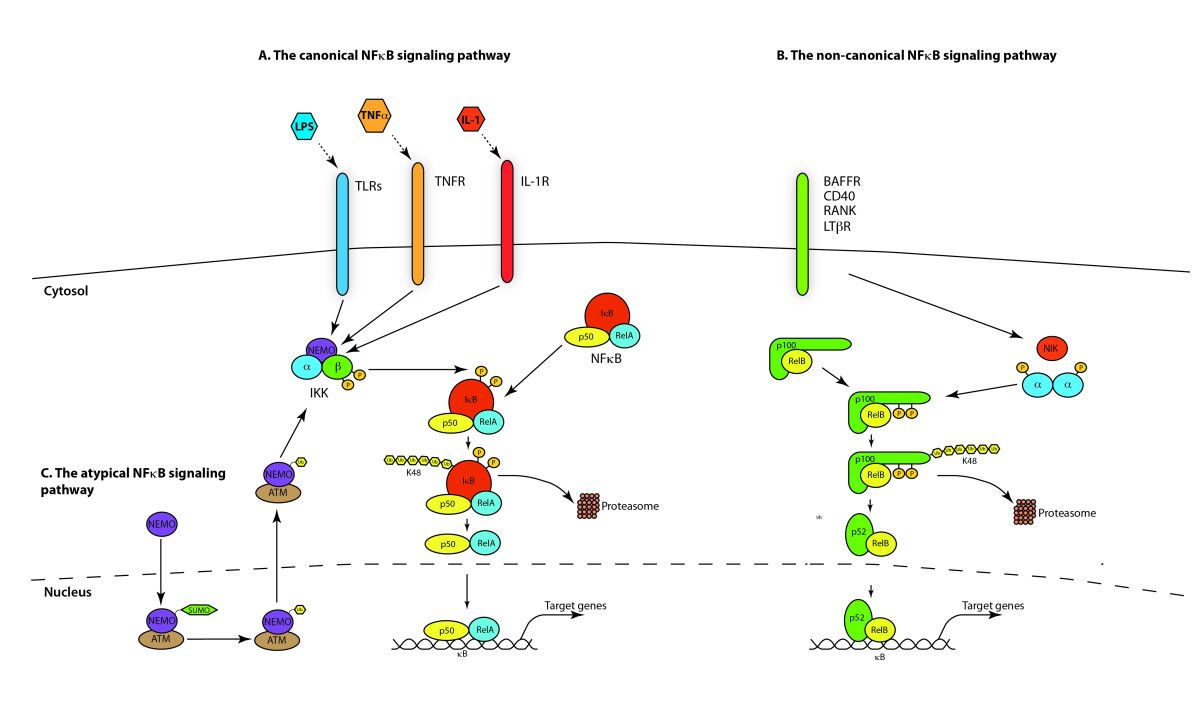
1.典型/经典级联
从促炎性细胞因子的细胞表面受体和病原体相关分子模式(PAMP),例如肿瘤坏死因子受体(TNFR),toll样受体(TLR)和T /
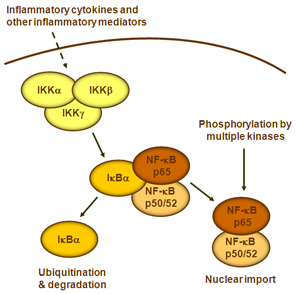
B细胞受体开始,进行规范或经典的级联信号传递。这些受体与其配体分子结合,并在细胞膜上传递信号,从而激活IkB激酶(IKK)复合物。该复合物的最常见形式包括IKKα和IKKβ催化亚基的异二聚体和IKKγ调节亚基。
IKKγ单元也称为NEMO,用于NF-kB基本调节剂。活化的IKK复合物主要以IKKγ依赖的方式通过IKKβ起作用,催化IkB的磷酸化(相当于IkBa的Ser32和Ser36的位点),多聚泛素化(相当于IkBa的Lys21和Lys22的位点)并随后通过26S蛋白酶体。释放的NF-kB二聚体(最常见的是p50–RelA二聚体),易位至细胞核,结合DNA并激活下游基因转录。
2.非规范/替代路径级联
另一个NF-kB激活途径称为非经典途径或替代途径。该途径独立于IKKβ和IKKγ,但依赖于IKKα调光器。信号通过LT-β或BAFF受体转移到细胞质中。使NIK蛋白磷酸化,然后使NIK蛋白磷酸化IKKα同二聚体。该途径中IKKα同二聚体的靶标是NF-kB2
/
p100,其在两个C末端位点被磷酸化。这些位点的磷酸化对于将p100加工成p52至关重要,而p52也依赖于多泛素化和蛋白酶体降解。然而,与其导致IkBs导致的p100完全降解,不如磷酸化依赖的p100泛素化仅导致其抑制性C端半部降解。一旦C末端的一半降解,就会释放NF-kB(包含RHD的p52多肽)的N末端部分。由于p100的RHD最常与RelB相关,因此这种“替代”途径的激活导致p52–RelB二聚体的核易位。最后,二聚体与DNA结合并激活下游基因转录。
3.途径调控
泛素是一种76个氨基酸的蛋白质,在从酵母到人的所有真核生物中均高度保守并广泛表达。遍在蛋白的C-末端甘氨酸中的羧酸可以通过异肽键与另一种蛋白质上的赖氨酸的ε-胺共价连接。该过程称为“泛素化”。此外,遍在蛋白的C末端尾巴可以直接连接到另一遍在蛋白的N末端蛋氨酸上以形成线性聚遍在蛋白链。泛素化在调节NF-kB途径中起着至关重要的作用。在未刺激的细胞中,NF-kB与kB家族的抑制蛋白(IkB)结合并被隔离在细胞质中。刺激后,IkB被IkB激酶(IKK)复合物磷酸化,磷酸化的IkB随后被泛素化并被26S蛋白酶体降解,从而使NF-kB易位至细胞核,从而调节大量基因的表达。作为IKK复合体的调节亚基,NEMO被认为是将泛素化信号转导至IKK激活的关键因素。几个DUB充当IKK的关键负调节剂,以严格控制NF-kB的活化。研究最深入的DUB之一是A20,有人提出A20通过通过N端OTU域连接到RIP1的K63连接的多聚泛素去泛素化,并促进RIP1的K48连接的多泛素化来促进蛋白酶体降解来抑制NFkB的过度活化。
4.下游信令
NF-kB充当免疫和炎症反应的主要介质,并参与应激反应以及细胞增殖和凋亡的调节。各自的NF-kB靶基因可使生物体有效地应对这些环境变化。经典的NF-kB途径被多种炎症信号激活,导致多种炎症和先天免疫基因的协调表达。促炎性细胞因子IL-1b和TNF-a激活NF-kB,并响应NF-kB激活而诱导其表达,从而形成放大的前馈环。
NF-kB的替代途径导致p52–RelB二聚体的核易位,严格依赖IKKa同型二聚体,并被NIK激活LTbR,BAFF和CD40L。许多数据强烈表明,该替代途径在涉及次级淋巴器官发育和维持的基因表达中起着核心作用。
NF-kB家族的转录因子响应环境变化而控制大量靶基因的表达,从而有助于协调炎症和免疫反应。
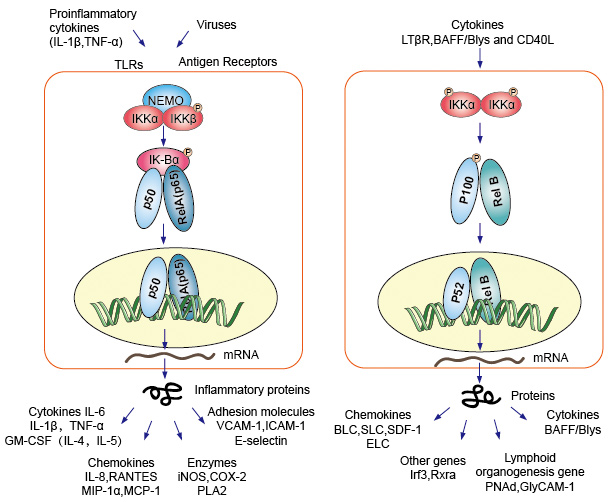
Figure 4. Down-stream signaling of NF-kB signaling pathway.
5.与疾病的关系。
代谢性疾病的炎症基础。
IKK /
NF-kB信号通路是代谢,炎症和胰岛素作用之间联系的关键。无论是由细胞内还是细胞外线索引起的导致胰岛素抵抗或胰腺β细胞功能异常的大多数代谢应激信号都集中在NF-kB激活激酶IKKb和其他主要的炎性激酶JNK-促分裂原活化蛋白激酶(MAPK)上。
NF-kB和糖酵解。
糖酵解可以比氧化磷酸化产生更多数量的ATP,并且生成速率更快。因此,葡萄糖是癌症和正常增殖细胞的必需营养素。
NF-kB协调许多在免疫,炎症和致癌过程中驱动细胞活化和增殖的信号。
NF-kB与氧化代谢有关。
NF-kB通过控制糖酵解和呼吸之间的平衡来控制能量的动态平衡和代谢适应。通过多种方式抑制NF-kB / RelA,可减少耗氧量并导致重编程为有氧糖酵解
The NF-kB
Signaling Pathway - Creative Diagnostics
https://www.creative-diagnostics.com/The-NF-kB-Signaling-Pathway.htm
NF-κB Signaling
Pathway
Nuclear factor-κB (NF-κB) belongs to a family of transcription factors that play
pivotal roles in inflammatory responses and immunological reactions. NFκB
pathway can be activated by a variety of stimuli, including TNF-α (tumor
necrosis factor α), interleukin 1 (IL-1), T and B cell mitogens, bacterial
lipopolysaccharide (LPS) and viral proteins. DiscoveRx offers a simple one-step
IκB degradation and NFκB translocation assay to study NFκB pathway. These fully
optimized, target-validated cell lines provide you convenience, relevant hits
and guaranteed performance for screening, profiling or research applications.
NF-κB信号通路
核因子-κB(NF-κB)属于转录因子家族,在炎症反应和免疫反应中起关键作用。
NFκB途径可被多种刺激激活,包括TNF-α(肿瘤坏死因子α),白介素1(IL-1),T细胞和B细胞有丝分裂原,细菌脂多糖(LPS)和病毒蛋白。
DiscoveRx提供了一个简单的一步式IκB降解和NFκB易位测定法来研究NFκB途径。这些经过完全优化,经过靶标验证的细胞系为您提供便利,相关的点击量,并为筛选,分析或研究应用提供有保证的性能。
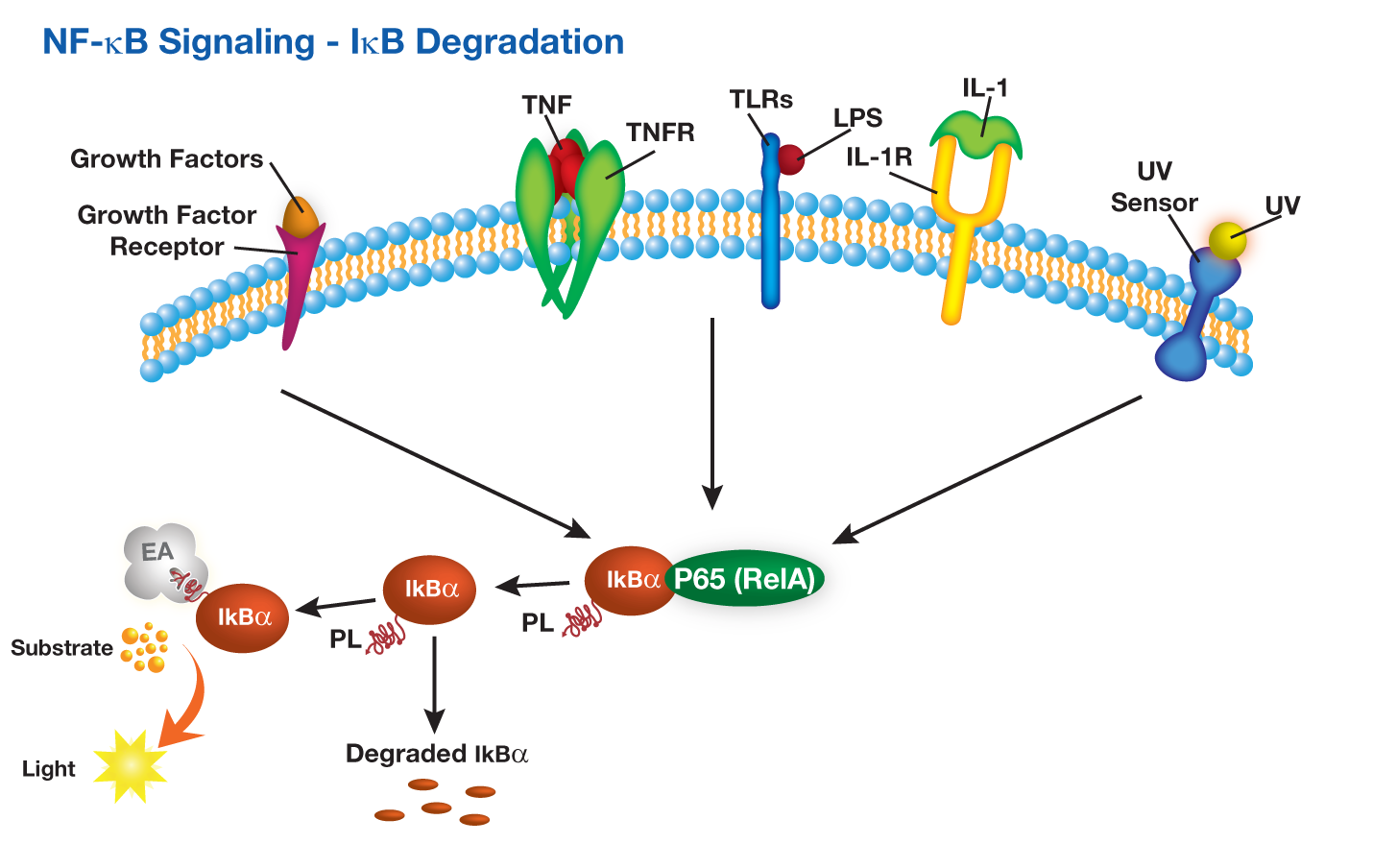
DiscoveRx提供PathHunter®IkB功能检测,其中用小的ProLabel肽标记IkB(κB抑制剂)。该测定法测量响应NFkB途径激活的人IκB降解。在TNFα或其他化合物存在下,细胞通过下游IκB蛋白发出信号。响应复合刺激,IκB蛋白降解。 PathHunter®IκB功能测定与PathHunter®PK / PL检测试剂盒(93-0180)一起使用。
DiscoveRx offers PathHunter® IkB functional assays in which IkB (Inhibitor of κB)is tagged with small ProLabel peptide. The assay measures human IκB degradation in response to activation of the NFkB pathway. In the presence of TNFα or other compounds, cells signal through downstream IκB proteins. IκB protein is degraded in response to compound stimulation. The PathHunter® IκB Functional Assay is used with the PathHunter® PK/PL Detection Kit (93-0180).
.png)
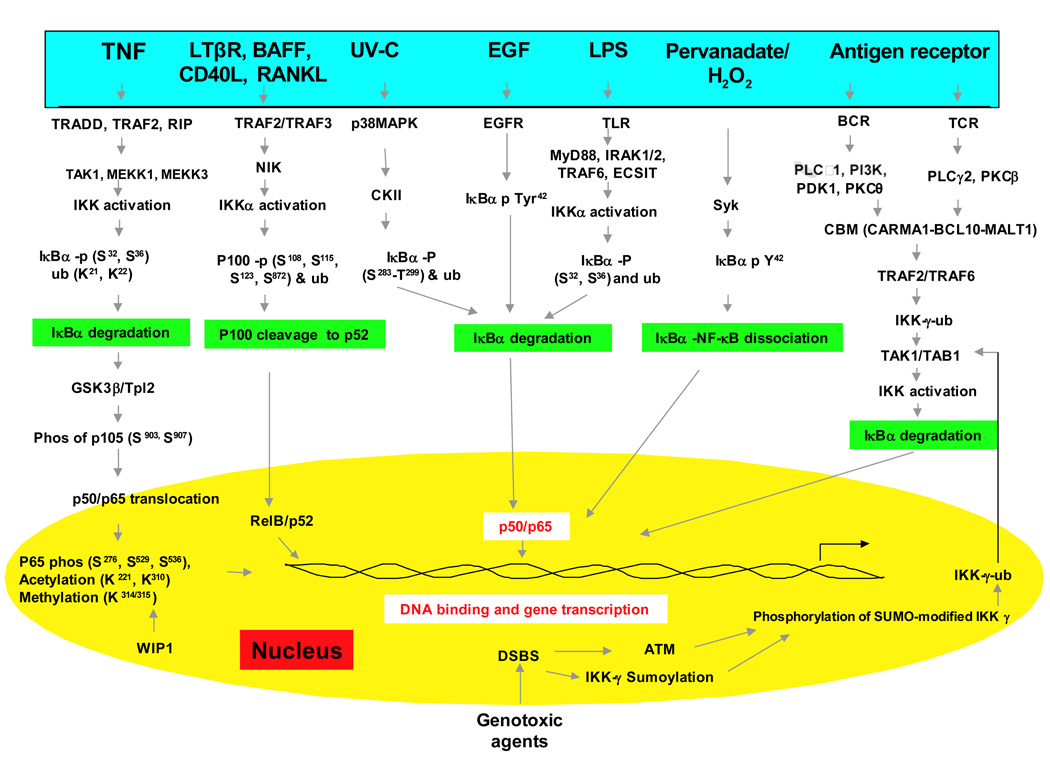
NF-κB激活主要通过其定位受到严格调控。在静息细胞中,NF-κB蛋白与抑制性IkB蛋白(包括IκBα,IκBβ和IκBε)相关联,其中IκBα最丰富。 NF-κB信号通过NF-κB1(p50 / p105)启动的经典(经典)途径和NF-κB2(p52 / p100)启动的非经典(替代)途径发生(图1)。在将活性NF-κB转运到细胞核之前,先将NF-κB1和NF-κB2裂解为活性p50和p52亚基。虽然经典途径依赖于由IKKα,IKKβ,IKKγ和抑制性亚基IκBs组成的IKK复合物,但替代途径依赖于IKKα同型二聚体和NF-κB诱导激酶(NIK)。在经典激活过程中,IKK复合物特异性磷酸化了两个保守的N端丝氨酸残基上的IκB,这些残基以E2和E3连接酶介导的多泛素化以及随后的26S蛋白酶体介导的降解为目标。这个过程释放并激活了NF-κB,后者现在易位至细胞核。通常与RelB相关的替代途径的激活导致p100前体蛋白向p52的加工受到调控,随后p52-RelB异二聚体向核移位[18]。尽管NF-κB激活主要通过经典和非经典途径发生,但在过去十年中,已经阐明了许多NF-κB激活途径(图1)。
Inhibiting NF-κB Activation by Small Molecules As a Therapeutic Strategy
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2955987/
![]()
“生存还是毁灭”细胞凋亡中调节NF-kB的结构性方式
1. “To be or not to be” A structural approach to the regulation of NF-κB in
Apoptosis By Rajendra
apoptosis
https://www.slideshare.net/RajKothinti/apoptosis-45768624
调节HIF1α和NFkB在癌症治疗中的作用:这是控制恶性进展的合理方法吗?
Modulators of HIF1α and NFkB in cancer treatment: is it a rational approach for controlling malignant progression?
NF-κB介导的炎症通路在细胞转化,癌症存活,增殖,侵袭,血管生成和转移中的作用。
NF-κB[核因子-κB]通过诱导炎症反应而在炎症中起重要作用。
NF-κB的活性与动脉粥样硬化,关节炎,炎性肠病,类风湿性关节炎和败血性休克,恶性转化和肿瘤生长(例如某些血液癌和实体瘤)以及骨分解和重建(骨质疏松症)有关。
NF-κB通常被认为是一种“生物开关”,可以在细胞中关闭以治疗这些疾病。
“NF-κB被多种因子激活,包括细胞因子,氧化剂自由基,吸入颗粒,紫外线辐射以及细菌或病毒产物。
研究机构:
1意大利罗马萨皮恩扎大学实验医学系
2分子和细胞病理学实验室–意大利罗马圣拉斐尔·皮萨纳科学技术研究所(Istituto di Ricovero e Cura a Carattere
Scientifico San Raffaele Pisana)
3意大利里贾纳埃琳娜·卡里斯特科学技术研究所外科病理学系,意大利罗马
4美国马里兰州巴尔的摩市约翰霍普金斯大学医学院细胞工程研究所血管计划
5美国夏威夷大学檀香山夏威夷大学夏威夷大学癌症中心约翰A.伯恩斯医学院
意大利罗马圣菲利波内里医院病理解剖学6UOC
7美国加利福尼亚州斯坦福市斯坦福大学医学系内分泌学,老年病学和代谢科
8意大利罗马Consiglio Nazionale delle Ricerche分子生物学与病理研究所
实际上,HIF1α和NFkB共同调节了上千个基因的转录,这些基因又控制着重要的细胞过程,例如适应缺氧,代谢重编程,炎症修复反应,细胞外基质消化,迁移和侵袭,粘附等。在这种广泛参与下,他们可以以综合的方式控制恶性表型的起源。有趣的是,通过发现警报蛋白受体基因(如RAGE,P2X7和某些TLR)被HIF1α激活,从而在肿瘤中相继弥合了缺氧和炎症。因此,警报蛋白受体又能强烈激活NFkB和促炎基因表达,证明了恶性表型的所有特征。最近,已经鉴定出许多药物,它们在抑制肿瘤进展方面抑制一种或两种转录因子,并具有令人鼓舞的结果。另外,这些分子中的许多是天然化合物或已经用于治疗其他病理的脱标签药物。其中一些正在接受临床试验,不久将被单独使用或与标准抗肿瘤药物联合使用,以减少转移形成,更重要的是净增加生存率,从而更好地治疗肿瘤。这篇综述强调了在肿瘤缺氧区域激活的HIF1α,转化细胞中NFkB激活和促炎基因表达的核心作用,以了解其向恶性肿瘤的进展。将审查抑制这些转录因子的不同分子和策略。最后,将概述一类称为Sirtuins的新型脱乙酰基酶在调节HIF1α和NFkB活性中的核心作用。
简介:癌症发病机制的新范式
当前关于癌症发病机理的流行理论仍然假设,致癌和转移的基本事件是单个细胞在其一生中积累的突变。尽管温伯格证明了在正常细胞中转染的突变癌基因和/或抑制基因的组合可以产生完全转化的细胞,但没有证据表明另一组基因可以产生恶性表型,侵袭并形成转移。
Hanahan和Weinberg(2011)提出,微环境可以以多种方式参与疾病的发展:提供VEGF,细胞因子以及其他生长和存活因子,主要来自活化的间充质和炎性细胞。并创造出富含活性氧(ROS)的微环境,这可能有助于新的突变。
转化突变
转化的特征是失去对增殖和/或凋亡的控制,并且其归因于与细胞周期有关的基因家族的功能增强(癌基因)或功能丧失(癌抑制基因)突变的积累控制和凋亡控制(Hanahan和Weinberg,2011;
Larsson,2011)。属于DNA修复机制的基因突变可能是该序列中上游步骤的原因,增加了具有转化表型所需的突变积累的机会。转化的实验模型无疑已经确定了这些基因的某些突变与转化表型的精确生成之间的因果关系(Elenbaas等,2001;
Ince等,2007)。
基因组不稳定性,化学诱变剂和辐射是可能导致涉及转化相关基因的随机突变的原因。与经典突变类似,表观遗传变化(DNA的甲基化或乙酰化状态)和染色质结构维持机制的改变可以稳定地实现对增殖和凋亡控制的生物学效应(功能获得,功能丧失)(Huang等(2011年;范登·伯格(Vanden
Berghe),2012年)。突变可以在生命的任何时期在体(干)细胞中建立,也可以存在于从亲本遗传的合子中。最后一种突变负责遗传性肿瘤风险,通常被认为是抑制癌基因功能的丧失。仅在异常情况下才能观察到癌基因(例如ret癌基因)的功能获得(Traugott和Moley,2010年)。这可以通过以下事实来解释:致癌基因突变的存在会破坏正常的形态发生和发育,从而导致胚胎或胎儿过早死亡。
适应进展的反应
进展的特征在于导致临床上显着的肿瘤的恶性表型的获得。恶性肿瘤包括在没有新形成的血管(新血管生成)的情况下生长到超出氧气和营养素扩散条件的有限尺寸的能力,使整个分子家族挤出和/或失活的能力(对药物的抵抗力),对邻近组织的侵袭(降解) BM和ECM),与原始组织分离的能力(黏附分子和性质的变化),对趋化因子(趋化因子和其他趋化因子的受体)的响应而迁移,在具有新肿瘤的特定部位归巢的能力(表达新的粘附分子集,这些粘附分子将在其他情况下被激活的远端内皮上遇到它们的受体; Furuta等,2010; Zigler等,2010; Noman等,2011; Nasr和Pelletier,2012)。这些基因中的大多数已经过单独研究和分析,以确定其突变,表观遗传变化和其他异常情况,以确定它们对恶性肿瘤的贡献。 然而,就所有必需基因的突变而言,了解恶性细胞的进程和所有特性一直令人失望和不切实际。这些基因是如此之多,以致于统计上不可能或不可能在整个人类生命中随机发生其突变。今天,人们普遍认为,尽管与进展相关的基因突变可能导致恶性肿瘤,但其他因素(不一定是突变)是导致恶性表型的致病序列的原因。
在最近的十年中,对肿瘤起源和表现出来的组织环境进行了深入研究。该分析的结果表明,宿主组织和肿瘤组织的微环境在许多方面都有助于肿瘤的进展和最终的形成。大量论文表明,这种贡献取决于所涉及的细胞,细胞之间的局部相互作用,所产生的旁分泌信号,局部低氧水平,是否存在活化白细胞活跃的局部免疫炎症反应以及许多其他
因素(Zigler等,2010;
Noman等,2011; Coleman等,2012; Hanahan和Coussens,2012; Hao等,2012; Mucaj等,2012;
Muratori和Tamagnone,2012; Nasr和Pelletier,2012年)。
在如此众多的不同贡献中,很难评估每种因素的确切作用以及它们在恶性肿瘤发生途径中的位置。此外,它们太异质,以致于不能试图全面解释恶性表型的各个方面而被包括在逻辑和顺序路径中。任何可用的统一框架,包括Hanahan和Weinberg(2011)最近提出的框架,都无法包含所有异类的观察和实验。
但是,来自许多实验室的论文趋向于统一解释早期转化细胞的进程。一方面,已经证明,基因适应低氧可以极大地促进恶性肿瘤的发展(Shay and
Celeste Simon,2012)。另一方面,已经表明许多促炎基因被恶性细胞过度表达(Tafani等,2011a,b; Jin等,2012;
Schito等,2012; Yao等,2012; De Santis等人,2013年)。桥接缺氧适应和癌细胞促炎基因表达,我们提出了以下假设:这两个非常复杂的细胞反应在依次激活时可能是解释恶性表型所有特性的良好候选框架。另外,我们建议转化的(仍未进行的)肿瘤干细胞最适合产生这种反应。
在下一段中,我们将分析低氧对转化癌细胞的分子和生物学效应。
癌症进展中的缺氧和炎症。在生长中的早期肿瘤中产生低氧和促炎性微环境
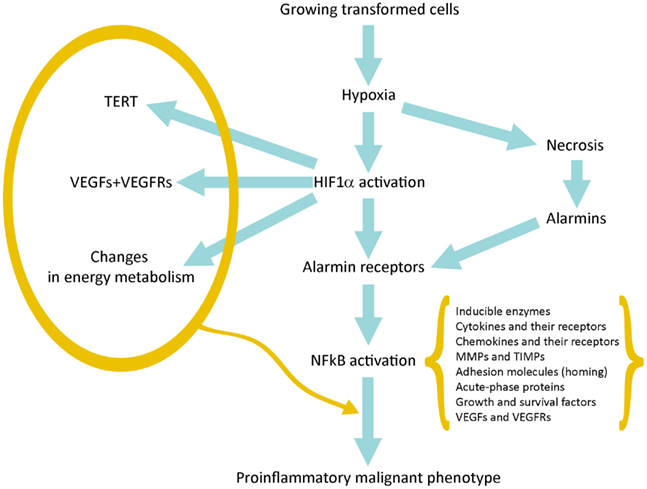
在没有新血管生成的情况下,早期转化的癌细胞能够增殖并形成小肿瘤。氧气和养分可以从宿主正常组织血管中扩散,半径不超过200μm(Brahimi-Horn等,2007)。当小肿瘤的直径超过400μm时,就会产生低氧环境,尤其是在肿瘤中心(Toffoli和Michiels,2008年)。
缺氧产生两个基本结果:(a)与宿主组织血管距离较远的细胞坏死; (b)HIF1α在更靠近血管并被严重破坏的存活肿瘤细胞中的激活;
HIF1α驱动的基因表达使它们得以生存和成长,从而增强了其发展到恶性肿瘤的承诺(图1)。
图1
图1 连接缺氧、HIF1α激活、炎症和肿瘤恶化的示意图。
Figure 1. Schematic representation of the pathway linking hypoxia and HIF1α activation with inflammation and tumor progression.
HIF1α的激活导致数百个基因的表达(表1)。它们中的许多提供了对肿瘤进展的第一冲动(承诺)。 VEGF及其受体负责新生血管生成,并有可能生长到直径超过400μm的极限。端粒酶激活增加了增殖潜力和可能的周期数;中间和能量代谢的变化是这种适应的最著名的代谢作用(Brahimi-Horn等,2007; Mucaj等,2012)。
.png)
坏死性损伤包括质膜碎裂和细胞内分子的释放,其中一些包括了警报蛋白(alarmin)或损伤相关分子模式(Damage-Associated
Molecular Patterns ,DAMP)。释放的警报蛋白与其受体(alarmin
receptors)的相互作用会触发多种细胞类型的促炎基因表达:常驻先天免疫细胞或白细胞,它们通常在其质膜上表达许多警报蛋白受体(alarmin
receptors)和肿瘤细胞,这些肿瘤细胞上表达了缺氧诱导产生的警报蛋白受体( Tafani等,2011a)。 警报蛋白受体(alarmin
receptor)信号传导导致NFkB激活,然后导致促炎基因表达。这种促炎性微环境可以促进肿瘤进展(见下文)。
关于HIF1α和HIF1α依赖基因的作用
通过检查由HIF1α激活的基因的功能,很明显,这些基因或基因家族中的许多在将转化细胞推向获得许多恶性肿瘤的标志中起着关键作用。尤其是,VEGF及其受体的过度表达(Ahluwalia和Tarnawski,2012)激活了肿瘤特异性的新血管生成,从而使早期肿瘤在氧气和养分的简单扩散作用下生长至200-300μm。端粒酶(TERT)的激活增加了端粒的长度和增殖潜能,使涉及的肿瘤细胞永生化(Guan等,2012)。典型的增生性基因(例如c-myc和cyclin
D1)的HIF1α依赖性激活为增生潜力做出了进一步贡献(Zhu等人,2010;
Fer和Melillo,2011)。此外,HIF1α激活OCT4和Notch促进干细胞更新,从而促进永生化(Lee等,2012;
Qiang等,2012)。对ABC转运蛋白的过度表达可实现对化学疗法的抵抗力(Maugeri-Saccà等,2011)。许多关键分子如ALDA,PGK,GLUT-1的过表达很好地解释了肿瘤能量代谢的重编程(葡萄糖转运和消耗增加以及乳酸产生的高糖酵解;
Semenza等,1996; Lam等)等人,2009; Mucaj等人,2012)。
HIF1α和NFkB共同控制着大多数侵袭和转移基因。因此,将在下一段中对其进行研究。
最后,更重要的是,我们观察到在低氧环境中,许多细胞类型(包括癌症和正常干细胞)从头表达或过表达不同的警报蛋白受体(类似于活化白细胞或CD45
+细胞中存在的那些; Tafani等。
,2011a)。在坏死细胞释放的警报蛋白激活后,RAGE,P2X7,TLR等会收敛于具有强大促炎基因表达的NFkB激活(图1)。这代表了关键的事件,它通过与数百种与炎症修复反应(Inflammation
Reparative
Response,IRR)相关的基因的表达来桥接对缺氧的适应性,并且非常重要的是,它获得了对恶性表型的经典特性。表1总结了主要参与低氧适应的恶性进展的基因或基因家族。该图还包括所谓的EMT(上皮-间质转化),其中所有涉及的基因都可能是HIF1α和/或NFkB依赖性的(Micalizzi等,2010)。
关于NFkB和NFkB依赖基因的作用
一旦通过许多不同的途径激活了NFkB,就会发生复杂的基因反应,数百个属于特定基因家族的基因的表达,包括与炎症和修复反应功能相关的大量成员(参见表2)。个别地,这些基因中的大多数已牵涉到恶性表型的关键特性的获得,从而提供了一个连贯的理论框架来解释大多数恶性标志的获得,作为整合反应和对肿瘤环境的适应。
表2. IRR基因表达和恶性表型的生物学特性。

诱导酶(COX2; 5-LOX,iNOS)
激活NFkB后在激活的白细胞中产生的可诱导酶负责产生介体分子,例如前列腺素,白三烯和NO,从而导致IRR的表现和扩增。它们在肿瘤微环境中的存在及其在肿瘤细胞自身中的表达一直是在癌症发病机理及其进展中涉及炎症的最早观察结果之一(Wang
and
Dubois,2006)。这些酶产生的分子有助于肿瘤进展的许多方面,例如新血管生成,白细胞募集到肿瘤微环境以及EMT的改变(Micalizzi等,2010)。大约15年前,一项具有里程碑意义的流行病学研究表明,使用小剂量阿司匹林预防心血管疾病可大大降低患结肠癌的风险(Gustafson-Svärd等,1997)。这些流行病学观察结果激发了许多其他回顾性研究,并进行了阿司匹林和其他COX2抑制剂在预防肿瘤及其进展方面的对照临床试验,从而开辟了了解炎症在肿瘤发病机制中作用的新时代。
细胞因子及其受体
细胞因子的特征是直接影响靶标白细胞的IRR,将反应极化为Th1或Th2并刺激靶细胞(CD45
+)增殖以增强和扩增IRR(DiDonato等人,2012)。细胞因子存在于大多数人类肿瘤微环境中,由癌细胞本身和/或白细胞浸润产生(DiDonato等,2012)。有趣的是,肿瘤细胞还表达与其恶性程度平行的各种细胞因子受体(DiDonato等,2012)。因此,由于细胞因子受体的存在,肿瘤细胞可以在生物学上受到很大的影响,例如增殖率(IL-2)和极化(Th1细胞因子),以及粘附分子及其抗受体的表达,从而影响转移的归巢(DiDonato等,2012)。
MMP和TIMP
MMP和TIMP是通常在活化白细胞中表达的NFkB依赖基因,但众所周知,MMP /
TIMP活性比的破坏,伴随基底膜和细胞外基质蛋白酶活性的增加存在于恶性肿瘤中并与侵袭性平行(Tobar等,2010;
Choi等,2011b)。然后,破坏这些生理组织屏障(极限)并开始入侵事件的开始,基本上是通过HIF1α和NFkB的表达而控制的。
黏附分子和反受体
白细胞中NFkB的激活可以很好地重新编程粘附分子的表达,以进行迁移并在组成区域组织或受损部位归巢。粘附分子的NFkB依赖性和/或细胞因子依赖性新表达也在肿瘤细胞中发生,从而允许许多通常与恶性肿瘤有关的生物学改变。这些变化包括脱离原始组织(即钙粘着蛋白)的能力,遵循特定趋化梯度和ECM分子路径(趋化因子和整联蛋白的受体)迁移的能力,以及最后归巢位点的确定由活化的内皮细胞代表(ICAM-1,选择蛋白及其反受体;
Marcu等人,2010)。
趋化因子及其受体
肿瘤细胞同时表达趋化因子及其受体,并与其恶性程度平行(Lu and
Kang,2010)。趋化因子的产生引起梯度,这可能是导致晚期肿瘤中白细胞的吸引和单核浸润的主要原因(Lu and
Kang,2010)。更重要的是,趋化因子受体的表达是转移发生的关键事件。实际上,转移是一个复杂的事件,包括数百个基因参与的多个步骤。从原发性肿瘤组织分离后,必须沿着趋化梯度进行矢量迁移,这意味着存在趋化因子特异性受体。
CXCR4是SDF1α的受体,在肿瘤细胞中得到最充分的表征,并且已与许多人类肿瘤的转移进展和转移预见相关(Lu和Kang,2010)。趋化因子及其受体都在NFkB的控制下。
VEGF和VEGFR
当前的成像技术可检测到的临床相关肿瘤,需要生长到直径几毫米的尺寸,然后默认情况下,它需要新血管生成过程,并在肿瘤微环境的血管中充分表达VEGF和VEGFR。
VGEFs可以由肿瘤微环境中存在的活化白细胞和间充质细胞产生,或更重要的是,在活化HIF1α和NFkB的影响下,肿瘤细胞本身也可以产生VGEF(Ono,2008)。在最后一种情况下,已经证明癌细胞(可能是肿瘤干细胞和祖细胞)也可以表达VEGFR,这表明肿瘤细胞可能有助于其新血管树的形成(Ono,2008)。
生长和生存因素
HIF1α和NFkB控制许多生长和生存因子及其受体。这已在活化的白细胞(参与组织修复)和缺氧活化的肿瘤细胞中得到证实。这是肿瘤生长的另一个优点,并且是建立继发转移性肿瘤的前提。
“种子和土壤”假说预示着有利的组织环境与转移的发生有关(Langley and
Fidler,2011)。在这种情况下,生长和存活因子既可以由微环境中活化的白细胞或间充质细胞提供,也可以由已经激活了增殖途径(转化癌基因)或已经被NFkB激活过表达这些基因的肿瘤细胞本身提供(Brahimi-Horn)等人,2007)。
急性期蛋白
急性期蛋白被认为是血浆标志物,可用于评估全身IRR。它们包括可溶性和细胞结合的同工型,例如C反应蛋白,pentraxin-3和其他pentraxin;它们的功能只是部分阐明。与其他NFkB依赖基因相似,它们在缺氧激活的肿瘤细胞和激活的白细胞中似乎表达或过表达。它们在肿瘤进展中的功能仍存在争议。一方面,它们似乎抑制了肿瘤细胞的增殖并随着进展而降低(Ronca等人,已提交),另一方面,与宿主正常组织相比,它们可以在恶性细胞中高表达(Hiratsuka等人,2008年)。
)。
SOCS和负调节器
NFkB激活还包括许多IRR负调控因子蛋白的表达,例如SOCS-1(Strebovsky et al。,2012)。后者是SOCS家族的成员,其通过JAK /
STAT抑制细胞因子信号传导,下调TLR表达和信号传导并降低NFkB活性和持续时间(Strebovsky等人,2012)。该系列和其他负调节器被认为是IRR正常反馈控制的一部分。正如我们的假设所预测的,由于HIF1α-NFkB整合激活的生理反应,SOCS-1在低氧激活的细胞中下降(De
Santis等,2013)。
抗HIF1α和抗IRR肿瘤治疗和癌症预防
许多流行病学研究和一些临床对照试验支持以下观点:IRR的负调节可降低某些肿瘤的风险和发生率,此外,还可以减慢或抑制其向恶性肿瘤的进展。这些观察结果和将缺氧、IRR和肿瘤进展联系在一起的实验研究提出了预防肿瘤,降低其发生率,减慢其进展,显著提高生存率和降低恶性肿瘤死亡率的新策略(Liu等人,2011)。
HIF和HIF依赖基因的调节剂
最近,由于其在肿瘤进展中的重要作用,HIF1α已成为越来越多开发的抑制剂的目标,这些抑制剂旨在阻止或减少肿瘤的生长以及可能的进展(Semenza,2012;
Xia等,2012;图2)。但是,必须注意,这些化合物中的大多数是FDA批准的用于治疗癌症或其他病理学或天然产物的分子,并且有关HIF1α抑制剂或激活剂的大多数研究和发现都是在基于细胞的系统或异种移植物中进行的由研究实验室而不是制药公司提供(Semenza,2012年)。迄今为止,已确定的抑制剂通过多种不同的机制发挥作用,这些机制包括降低HIF1α的mRNA和蛋白质水平,防止HIF1α二聚化或DNA结合,抑制HIF1α与共激活因子的结合。图3报告了一些在HIF1α途径不同步骤起作用的代表性化合物。

图2.HIF1α激活可在该途径的不同步骤被抑制
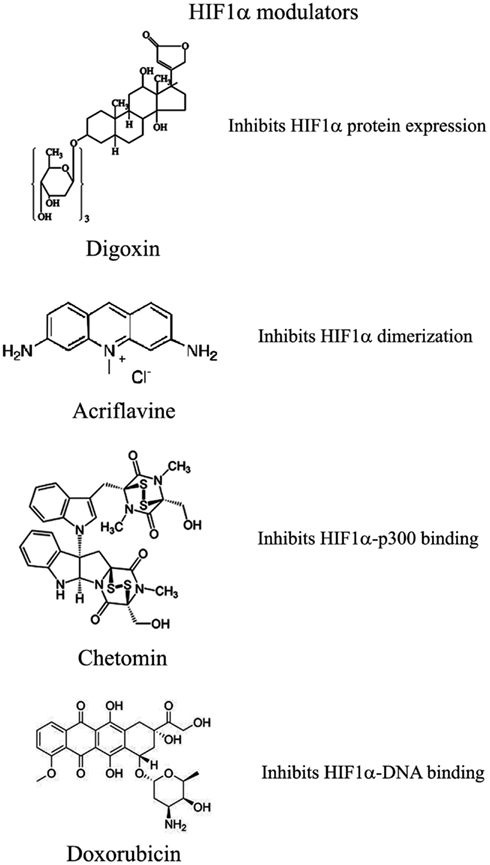
图3.可以用来调节HIF1α途径的化合物的选择
已显示几种化合物可减少HIF1α蛋白和mRNA的积累
其中:(1)PI3K激酶抑制剂渥曼青霉素和LY294002以及mTOR抑制剂雷帕霉素已显示可降低不同细胞系中的HIF1α蛋白水平(Jiang等,2001;
Majumder等,2004)。实际上,这些研究表明,mTOR对雷帕霉素敏感的功能对于HIF1α的积累不是必需的,但对于该蛋白的充分表达以及对氧气和营养不良条件的整合至关重要(Majumder等,2004)。
(2)地高辛,哇巴因等强心苷也获得了抑制HIF1α蛋白表达和降低异种移植瘤生长的作用(Semenza,2012)。地高辛已长期用于治疗心脏病,因此,与其他常规抗肿瘤药联合治疗尤为重要(Zhang等,2008)。
(3)微管靶向剂,例如2甲氧基雌二醇(2ME2)及其合成衍生物,可阻止HIF1α翻译和核蓄积,并具有相应的抗肿瘤活性(Mabjeesh等,2003)。然而,尚未完全阐明此类化合物的确切作用机理。
(4)II类组蛋白脱乙酰基酶(HDAC)抑制剂,例如曲古抑菌素和LAQ824,通过未知机制增加HIF1α的泛素化和HIF1α的降解(Qian等,2006)。下文将详细讨论III类HDAC在HIF1α稳定性方面的新兴作用。
(5)基于锁定核酸(LNA)的寡核苷酸是第三代反义技术,可提供高稳定性和持久的靶标抑制作用。
EZN-2968是针对HIF1α的LNA,已显示出对HIF1α和HIF1α依赖性基因的强烈抑制作用,并且由于其能够减少异种移植物中肿瘤的生长,目前正处于I期临床研究(Greenberger等,2008)。
(6)布洛芬和其他NSAID通过尚未确定可能涉及PI3K或蛋白酶体的机制降低了前列腺癌细胞中的HIF1α和HIF2α蛋白水平(Palayoor等,2003)。但是,布洛芬对HIF的降解并不是由于其对COX2的抑制作用(Isaacs等,2002)。
(7)天然抗生素格尔德霉素(GA)和抗真菌紫杉醇可防止Hsp90与HIF1α结合,从而降低其稳定性并随后发生蛋白酶体降解(Isaacs等,2002)。特别是,GA的衍生物目前正在临床试验中。同样,Antimycin
A,来自链霉菌属的一种抗生素。诱导细胞凋亡并抑制从细胞色素b到细胞色素C1的线粒体电子转运链,通过未知机制降低了HIF1α蛋白水平(Maeda等,2006)。(8)许多天然产物具有HIF1α的抑制作用。这些物质中的许多物质通过激活蛋白酶体系统或未知机制来增加HIF1α降解。尤其是桑树皮(桑树皮)产生的moracin
O 和 moracin P会激活HIF1α降解(Dat等,2009)。其他HIF1α抑制剂是水生植物金冠龙(Saururus
cernus)的金黄色素B,可能通过降解HIF1α和抑制VEGF分泌发挥其作用(Hossain等,2005)。姜黄素(curcumin)和黄连素(berberine)分别来自印度香料姜黄和中国金丝,可增加HIF1α蛋白酶体降解(Choi等,2006)。用白藜芦醇(一种在葡萄和其他植物中发现的化合物)以及类黄酮(例如热带豆科植物Lonchocarpus
glabrescens的甲基铝松黄酮)也获得了类似的结果(Park等,2007;
Liu等,2009)。红色鼠尾草中的西比里醌A通过HIF1α降解抑制HIF1α积累和VEGF分泌(Dat等,2007)。该部分清单清楚地表明,天然产物是HIF1α抑制剂的重要来源,其通过多种不同的机制起作用,其中许多机制仍然未知。
已通过两种化合物证明了HIF1α二聚体的抑制作用
Acriflavine和韩国红参。
Acriflavine是一种抗菌剂,可与HIF1α和HIF2α的PAS-B子域结合,从而阻止与HIF1β的结合,这种作用可降低VEGF的产生和肿瘤的生长(Lee等,2009)。红参水提取物抑制HIF1α和1β二聚化,没有毒性作用,但是其作用机理和抗肿瘤作用尚不清楚(Choi等,2011a)。
HIF与HRE的DNA结合以及HIF1α依赖基因的表达已被几种化合物抑制
与HIF1α识别的HRE元素结合并抑制其合成的聚酰胺会阻止VEGF的合成(Semenza,2012)。阿霉素和柔红霉素与DNA结合并阻止HIF结合,靶基因转录和肿瘤生长(Tanaka等,2012)。最后,棘轮霉素是一种从紫锥菊链霉菌中分离出来的抗生素,可与DNA结合并抑制HIF1α的活性(Wang等,2011)。
HIF1α转录活性需要与共激活因子p300结合
壳聚糖的一种代谢产物,化学发光蛋白,通过作用于p300结构来阻止HIF-p300结合并抑制HIF靶基因的转录(Kung等,2004)。在裸鼠中,Chetomin可在不影响体重的情况下阻止肿瘤生长(Kung等,2004)。蛋白酶体抑制剂Bortezomib结合到与p300相互作用的HIF1α域上,从而阻止了这两个因子之间的功能性相互作用并阻止了靶基因的转录(Befani等,2012)。
NFkB和NFkB依赖性基因的调节剂
转录因子NFkB在肿瘤发生过程中起着核心作用,因为它促进涉及关键细胞路径的500多个基因的表达,例如抑制凋亡,增加迁移和侵袭,增加细胞外基质的消化,增加粘附分子的表达等(Gupta等人,2010年;图4)。
NFkB受许多翻译后修饰的调控,例如甲基化,乙酰化,磷酸化,泛素化。此外,一旦被激活,NFkB就会在细胞核内转移并积聚,并与DNA结合并激活大量基因的转录(Gupta等,2010)。因此,类似于HIF1α,也可以通过作用于控制其活性的路径的不同步骤来调节NFkB。同样,许多主要具有抗炎特性的天然产物是NFkB抑制剂(Gupta等,2010)。迄今为止,已鉴定出700多种NFkB抑制剂,其重要性是由于该转录因子对许多炎症和癌症之外的病理的重要作用(Wilczynski等,2011)。图5报告了一些在NFkB路径不同步骤起作用的代表性化合物。
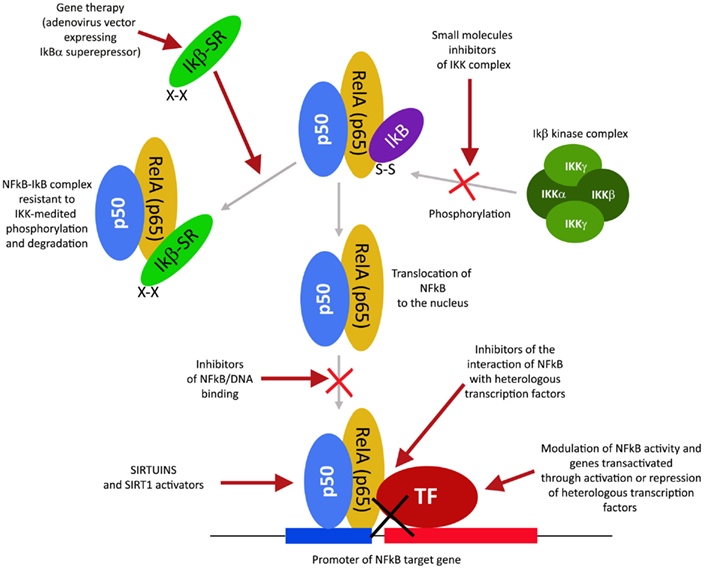
图4. NFkB激活途径及其不同的抑制步骤。

图5 图5.可以用于调节NFkB途径的化合物的选择。
www.frontiersin.org
此外,有可能通过考虑其作用机理将大量的NFkB抑制剂归类。尤其是:
通过蛋白激酶抑制或蛋白磷酸酶激活来调节NFkB
这类NFkB抑制剂可阻止IKK激酶的激活。
IKK磷酸化并增加抑制性亚基IkB的降解,从而激活NFkB(Gupta等,2010)。然而,仅对于少数IKK抑制剂,其作用机理是已知的(Gupta等,2010)。在这些IKK抑制剂中,有天然化合物,例如B-咔啉(Karin等人,2004),一种从几种植物中提取出来的吲哚生物碱,它起苯并二氮杂激动剂的作用,孤儿菊内酯,一种从白菊中分离出来的天然化合物,通过与IKK的Cys-179和姜黄素(Lubbad等,2009;
Gupta等,2010)。尤其是,单去酚内酯似乎也对癌症干细胞具有抗肿瘤活性(Gunn等,2011)。抗炎药是IKK抑制剂,尽管其机制尚不清楚(Wilczynski等,2011)。其中包括:阿司匹林,布洛芬,舒林酸,柳氮磺吡啶和其他非甾体抗炎药(卡斯特,2006年)。其它NFkB抑制剂是一些激酶抑制剂,例如SB203580,PD0980589,酪氨酸激酶抑制剂,甜菜碱等(betaine)等(Vanden
Berghe等,1998)。最后,腺病毒传递的IKK显性负激酶和抗IKK抗体也是可以抑制NFkB激活的替代策略(Gupta等,2010)。相比之下,NFkB可以通过激活减少IkB磷酸化的磷酸酶来抑制。例如,胞嘧啶阿拉伯糖苷使NFKB去磷酸化并诱导肿瘤细胞凋亡(Sreenivasan等,2003)。
蛋白酶体抑制调节NFkB
该策略利用了蛋白酶体系统抑制IkB降解导致NFkB抑制这一事实。充分研究和使用的蛋白酶体抑制剂是Bortezomib(也称为HIF1α抑制剂),已显示可减少异种移植模型中的肿瘤生长,并已成功与其他药物(如顺铂)联合用于一线治疗(Wilczynski
et al。
,2011)。硼替佐米抑制HIF1α,VEGF和肿瘤血管形成是伴随NFkB抑制的其他好处。其他与硼替佐米相似的蛋白酶体抑制剂包括ALLnL,乳腺嘧啶,MG132等。最后,第二代蛋白酶体抑制剂是卡非佐米和盐孢菌酰胺,它们在纳摩尔范围内起作用,毒性较低,可以口服给药(Wilczynski等,2011;羽衣甘蓝)和摩尔,2012年)。
通过乙酰化抑制调节NFkB
类似于激酶的磷酸化和磷酸酶的去磷酸化,乙酰基转移酶和脱乙酰基酶的乙酰化/脱乙酰基被认为是控制许多蛋白质活性的同等重要的翻译后修饰(Gray and
Teh,2001)。已经证明NFkB在几种赖氨酸上被乙酰化,这增加了其活化(Kiernan et
al。,2003)。因此,如以下部分中的主要细节所述,最近发现的一类名为Sirtuins的新型脱乙酰基酶已引起人们对通过这些酶控制NFkB活性的可能性的关注。实际上,sirtuins的激活会引起NFkB的抑制。同样,NFkB可以通过抑制乙酰转移酶(例如p300和CREB结合蛋白)来抑制(Chen和Greene,2004)。实际上,已显示来自坚果或橡树皮的没食子酸(gallic
acid)和熊果酸可抑制NFkB乙酰化并随后激活(Choi等,2009)。
通过抑制核积累来调节NFkB
该方法基于这样一个事实,即通过在细胞核中蓄积来预防NFkB也会阻止其DNA缔合和靶基因的转录。但是,这种机制仅针对SN50(具有疏水性膜转运区和NFkB的NLS)的肽进行了记录。
SN50与NFkB竞争核转运机制,从而防止NFkB核转运。不幸的是,SN50阻止了大量转录因子的核积累(Sun等,2012)。更有前景的抑制剂是衍生自真菌抗生素的化合物,称为脱羟甲基环氧喹诺酮(DHMEQ),可特异性抑制NFkB核积累,具有抗炎和抗肿瘤活性(Kozakai等,2012)。
Sirtuins和SIRT1激活剂调节HIF1α和NFkB
Sirtuins的名称归因于沉默信息调节剂2(Sir2),其在酵母中鉴定出并与寿命延长相关(Houtkooper等,2012)。在哺乳动物中,有七个Sir2同源物(SIRT
1-7)。 Sirtuins是III类烟酰胺腺嘌呤二核苷酸-(NAD
+)依赖性脱乙酰基酶或ADP-核糖基转移酶(Houtkooper等人,2012)。它们对NAD
+的依赖性直接将沉默调节蛋白的活性与细胞的代谢状态联系起来。因此,sirtuins参与了许多生理功能,例如基因沉默,细胞死亡,寿命,炎症和癌症(Houtkooper等,2012)。
Sirtuins还显示出缔合,脱乙酰基化并调节HIF1α和NFkB的活性。但是,仅针对SIRT1、2、3和6,该调节功能已得到证明。
SIRT1使HIF1α和NFkB脱乙酰。对于NFkB,到目前为止,所有结果均表明SIRT1脱乙酰后可抑制其信号传导(Morris,2012年)。实际上,体外和体内实验均显示SIRT1或白藜芦醇和其他多酚对SIRT1的激活通过使乙酰基化和抑制NFkB降低炎症反应。考虑到NFkB在涉及炎症,衰老,癌症等许多细胞途径中的核心作用,这些结果特别有趣。相反,已经报道了有争议的结果,涉及SIRT1
/HIF1α信号传导。实际上,尚不清楚SIRT1是否受缺氧影响。一些报道表明低氧会增加SIRT1水平,而其他报道则表明低氧会降低SIRT1(Lim等,2010;
Laemmle等,2012)。在缺氧条件下,SIRT1使HIF1α脱酰基,这种反应在某些情况下会降低HIF1α的活性,而在另一些情况下则会增加HIF1α的活性。显然,在描述SIRT1在HIF1α上的真正功能之前,必须通过在不同的细胞系、组织和体内模型上积累更多的数据。此外,HIF1α的SIRT1作用在不同的组织和器官中也可能不同。
SIRT6是另一个可控制HIF1α和NFkB乙酰化状态和转录活性的核沉默蛋白。在HIF1α的情况下,SIRT6充当HIF1α转录活性的核心抑制剂,使HIF1α靶基因启动子处的组蛋白3赖氨酸8(H3K9)脱乙酰。实际上,SIRT6调节葡萄糖通量显得至关重要,因为SIRT6缺乏会导致致命的低血糖症(Zhong等,2010)。有趣的是,SIRT6使用了类似的机制来抑制NFkB功能。同样在这种情况下,SIRT6使选定的NFkB靶基因启动子上的H3K9脱乙酰基化,从而降低了这些启动子对NFkB的可获得性(Kawahara等,2009)。重要的是,在缺乏SIRT1的小鼠中,SIRT6通过减弱由于乙酰化状态增加而增加的NFkB活性而显示出补偿作用(Schug等人,2010)。总之,SIRT1和SIRT6虽然具有不同的机制,但它们均代表NFkB活性的负调节剂。
已显示SIRT2使细胞质中赖氨酸310(K310)上的NFkB亚基p65脱乙酰化(Rothgiesser et
al。,2010)。这样,SIRT2在TNF刺激后抑制NFkB激活和NFkB靶基因的转录(Rothgiesser等,2010)。实际上,SIRT2沉默的细胞在TNF暴露后具有增强的NFkB激活和较低的细胞死亡百分比(Rothgiesser等,2010)。因此,NFkB可以被胞浆中的SIRT2和细胞核中的SIRT1脱乙酰。
SIRT3间接控制HIF1α的激活。实际上,SIRT3可以降低线粒体ROS,并激活细胞路径和清除ROS的酶(Finley等,2011;
Pellegrini等,2012)。特别是,通过降低ROS水平,SIRT3可以稳定HIF降解酶脯氨酰羟化酶(PHD),从而降低HIF1α的水平(Finley等,2011)。有趣的是,SIRT3缺乏与异种移植物中的肿瘤生长有关,并且在几种癌症和癌细胞系中,SIRT3的表达均降低(Finley等,2011)。
鉴于Sirtuins蛋白同时调节HIF1α和NFkB以及这两个转录因子在肿瘤进展过程中起着核心作用,因此为了控制HIF1α和NFkB而作用于瑟土因蛋白的可能性备受关注。因此,目前,在生产Sirtuins调节剂上已作了大量努力。几种天然化合物,例如白藜芦醇,槲皮素,皮卡三醇和其他多酚,已显示出调节sirtuins的功能,尤其是SIRT1的功能(Chung等,2010;
Gertz等,2012)。但是,它们的作用不仅限于SIRT1,还影响其他酶,例如磷酸二酯酶(PDE)和AMP激酶(AMPK; Dallas等,2008)。
sirtuins的抑制剂和活化剂的最新、准确的综述最近已发表(Villalba和Alcaín,2012年)。
正在进行的临床试验和未来方向
HIF1α
对HIF在肿瘤生长和进展中的核心作用的认识以及许多HIF1α抑制剂在体外和体内对肿瘤生长的抑制作用的认识,目前正在其中的某些临床试验中转化。特别是2ME2正在乳腺癌,前列腺癌和卵巢癌患者中进行II期临床试验(Semenza,2012;
Xia等,2012)。同样,还选择了2ME2衍生的分子用于I期临床试验的评估(Semenza,2012;
Xia等,2012)。格尔德霉素(GA)的类似物正在VHL疾病,乳腺癌等患者的II期临床试验中(Semenza,2012;
Xia等,2012)。根据II期试验的结果,硼替佐米(velcade)在美国已获FDA批准用于多发性骨髓瘤。两项开放性III期临床试验确定了硼替佐米1.3 mg
/ m2(有或没有地塞米松)对复发/难治性多发性骨髓瘤患者的疗效(Semenza,2012; Xia等,2012)。
EZN-2968目前处于I期临床试验中(Semenza,2012; Xia等,2012)。
最后,一个重要的考虑因素是,许多与HIF1α相互作用的药物正在临床癌症试验中或已被批准用于治疗癌症或其他疾病(Xia等,2012)。
NFkB
目前正在与化学疗法和放射疗法结合测试几类NFkB抑制剂。实际上,许多临床试验正在测试合理设计的抑制NFkB的药物的功效和特异性(有关在美国的特定试验,请参见NIH服务:ClincalTrials.gov)。例如在NFkB的情况下,蛋白酶体抑制剂Bortezomib(也用于抑制HIF1α)具有令人惊讶的有限副作用,因此,由于抑制作用可能引起的潜在不良反应,目前看来没有理由拒绝NFκB作为药物靶标转录因子的表达(Wilczynski
et al。,2011)。最近,在II期临床试验中,发现姜黄素对晚期胰腺癌患者有益(Wilczynski等,2011;
Grynkiewicz和Slifirski,2012)。这些只是目前正在测试的大量NFkB抑制剂的两个例子。希望在未来几年中,可以提高化疗和放疗治疗效率的几种NFkB抑制剂将成功用于癌症患者的治疗。
Sirtuins
考虑到这些酶的研究时间相对较短,因此很少对沉默调节蛋白的调节剂进行试验。实际上,目前,尚无可特异性调节单个沉默调节蛋白的调节剂。然而,白藜芦醇及其许多衍生物已在人类的随机双盲交叉试验中显示出有益的作用,其作用类似于卡路里限制和AMPK,SIRT1和PGC-1α水平的激活(Timmers等,2011
)。已提出将SIRT1和SIRT2的抑制剂用于治疗癌症,但远未进行临床试验(Morris,2012年)。
结论
考虑到HIF1α和NFkB在代谢重编程,炎症和癌症中的核心作用,并且还考虑到两个转录因子均受Sirtuins调节的事实,开发出作用于这些分子网不同步骤的更特异性调节剂的可能性取得了可喜的结果寻求更高成功率的新疗法。
缩写:
IRR:Inflammatory
Reparative Response (IRR)
rontiers | Modulators of HIF1α and NFkB in Cancer Treatment: Is it a Rational Approach for Controlling Malignant Progression? | Pharmacology https://www.frontiersin.org/articles/10.3389/fphar.2013.00013/full
HIF1α and NFkB are two transcription factors very frequently activated in
tumors and involved in tumor growth, progression, and resistance to
chemotherapy. In fact, HIF1α and NFkB together regulate transcription of over a
thousand genes that, in turn, control vital cellular processes such as
adaptation to the hypoxia, metabolic reprograming, inflammatory reparative
response, extracellular matrix digestion, migration and invasion, adhesion, etc.
Because of this wide involvement they could control in an integrated manner the
origin of the malignant phenotype. Interestingly, hypoxia and inflammation have
been sequentially bridged in tumors by the discovery that alarmin receptors
genes such as RAGE, P2X7, and some TLRs, are activated by HIF1α; and that, in
turn, alarmin receptors strongly activate NFkB and proinflammatory gene
expression, evidencing all the hallmarks of the malignant phenotype. Recently, a
large number of drugs have been identified that inhibit one or both
transcription factors with promising results in terms of controlling tumor
progression. In addition, many of these molecules are natural compounds or
off-label drugs already used to cure other pathologies. Some of them are
undergoing clinical trials and soon they will be used alone or in combination
with standard anti-tumoral agents to achieve a better treatment of tumors with
reduction of metastasis formation and, more importantly, with a net increase in
survival. This review highlights the central role of HIF1α activated in hypoxic
regions of the tumor, of NFkB activation and proinflammatory gene expression in
transformed cells to understand their progression toward malignancy. Different
molecules and strategies to inhibit these transcription factors will be
reviewed. Finally, the central role of a new class of deacetylases called
Sirtuins in regulating HIF1α and NFkB activity will be outlined.
Introduction: A New Paradigm on Cancer Pathogenesis
The currently prevalent theory on cancer pathogenesis still assumes that the
basic events for carcinogenesis and metastasis are mutations that are
accumulated by a single cell during its life. Although Weinberg demonstrated
that a combination of mutated oncogenes and/or suppressor genes transfected in a
normal cell can produce a fully transformed cell, there is no demonstration that
another set of genes can produce the malignant phenotype, invading and forming
metastasis.
Hanahan and Weinberg (2011) proposed that microenvironment could participate to
the progression in many ways: providing VEGF, cytokines, and other growth and
survival factors, mostly from activated mesenchymal and inflammatory cells; and
creating a reactive oxygen species (ROS)-rich microenvironment which could favor
new mutations.
Mutations for Transformation
Transformation is characterized by loss of control of proliferation and/or of
apoptosis and it is due to an accumulation of mutations with gain-of-function
(oncogenes) or loss-of-function (oncosuppressor genes) of gene families related
to the cell cycle control and apoptosis control (Hanahan and Weinberg, 2011;
Larsson, 2011). Mutations of genes belonging to the DNA repair mechanisms may be
responsible for the upstream steps in this sequence, increasing the chances of
accumulation of the mutations needed to have a transformed phenotype.
Experimental models of transformation definitely have established the
cause/effect relationship between certain mutations of these genes and the
precise generation of a transformed phenotype (Elenbaas et al., 2001; Ince et
al., 2007).
Genomic instability, chemical mutagens, and radiations are responsible for
random mutations that can involve transformation-related genes. Epigenetic
changes (methylation or acetylation status of DNA) and alterations in chromatin
structure maintenance mechanisms can stably achieve biological effects
(gain-of-function, loss-of-function) on proliferation and apoptosis control,
similar to the classical mutations (Huang et al., 2011; Vanden Berghe, 2012).
Mutations can be established in somatic (stem) cells in any period of life or
can be present in the zygote being inherited from parents. This last type of
mutations is responsible for inherited tumor risk and usually regards a
loss-of-function of oncosuppressor genes. A gain-of-function of oncogenes (such
as ret oncogene) can be observed only exceptionally (Traugott and Moley, 2010).
This can be explained by the fact that the presence of oncogene mutations,
disrupting normal morphogenesis and development, lead to premature embryonic or
fetal death.
In the next paragraph we will analyze the molecular and biological effects of
the hypoxia on transformed cancer cells.
Hypoxia and Inflammation in Cancer Progression. Generation of a Hypoxic and
Proinflammatory Microenvironment in a Growing Early Tumor
Early transformed cancer cells are able to proliferate and form small tumors in
the absence of neoangiogenesis. Oxygen and nutrients can diffuse from host
normal tissue vessels over a radius of no more than 200 μm (Brahimi-Horn et al.,
2007). When the small tumor reaches more than 400 μm in diameter, a hypoxic
environment is generated, especially in the center of the tumor (Toffoli and
Michiels, 2008).
Hypoxia produces two basic consequences: (a) Necrosis of cells that are more
distant from vessels of host tissue; (b) Activation of HIF1α in surviving tumor
cells closer to the vessels and sublethally damaged; the HIF1α-driven gene
expression allows them to survive and grow increasing their commitment to
malignancy (Figure 1).
FIGURE 1
www.frontiersin.org
Figure 1. Schematic representation of the pathway linking hypoxia and HIF1α
activation with inflammation and tumor progression.
Activation of HIF1α leads to the expression of hundreds genes (Table 1). Many of
them provide a first impulse (commitment) toward tumor progression. VEGFs and
their receptors are responsible for neoangiogenesis and for the possibility to
grow above the limit of 400 μm in diameter; telomerase activation increases the
proliferative potential and the number of possible cycles; and changes in
intermediate and energy metabolism are the best known metabolic effects of this
adaptation (Brahimi-Horn et al., 2007; Mucaj et al., 2012).
TABLE 1
www.frontiersin.org
Table 1. Adaptation to hypoxia in transformed (stem) cells.
Necrotic damage include plasma membrane fragmentation and release of
intracellular molecules, some of which constitute alarmins or Damage-Associated
Molecular Patterns (DAMPs). The interaction of released alarmins with their
receptors triggers a proinflammatory gene expression in various cell types:
resident innate immunity cells or leukocytes, which usually express in their
plasma membrane a number of alarmin receptors and tumor cells in which alarmin
receptors have been induced by hypoxia (Tafani et al., 2011a). Alarmin receptor
signaling leads to the activation of NFkB and then to the proinflammatory gene
expression. This proinflammatory microenvironment can contribute to tumor
progression (see below)
Table 2. IRR gene expression and malignant phenotype biological properties.
Inducible enzymes (COX2; 5-LOX, iNOS)
Inducible enzymes produced in activated leukocytes upon activation of NFkB are
responsible for mediator molecules production such as prostaglandins,
leukotrienes, and NO, leading to the manifestation and amplification of the IRR.
Their presence in tumor microenvironment and their expression by tumor cells
itself has been one of the earliest observation involving inflammation in the
pathogenesis of cancer and its progression (Wang and Dubois, 2006). Molecules
produced by these enzymes contribute to the many aspects of tumor progression
such as neoangiogenesis, recruitment of leukocyte to the tumor microenvironment,
and changes for EMT (Micalizzi et al., 2010). Almost 15 years ago a landmark
epidemiological study suggested that the use of low-dose aspirin for
cardiovascular prevention drastically reduced the risk for colon cancer
(Gustafson-Svärd et al., 1997). These epidemiological observations stimulated a
number of other retrospective studies and controlled clinical trials on aspirin
and other COX2 inhibitors in preventing tumors and their progression, giving
rise to a new era in the understanding the role of inflammation in tumor
pathogenesis.
Cytokines and their receptors
Cytokines characterizes IRR directly influencing target leukocytes, polarizing
the response as Th1 or Th2 and stimulating the proliferation of target cells
(CD45+) to reinforce and amplify the IRR (DiDonato et al., 2012). Cytokines are
present in most human tumor microenvironment, being produced by cancer cells
itself and/or by leukocyte infiltrate (DiDonato et al., 2012). Interestingly,
tumor cells express also receptors for various cytokines in parallel with their
degree of malignancy (DiDonato et al., 2012). Therefore, thanks to the presence
of cytokine receptors, tumor cells can be strongly influenced in their biology,
such as proliferation rate (IL-2) and in their polarization (Th1 cytokines) and,
probably, in the expression of adhesion molecules and their countereceptors,
thus influencing the homing for metastasis (DiDonato et al., 2012).
MMPs and TIMPs
MMPs and TIMPs are NFkB-dependent genes normally expressed in activated
leukocytes, but it is well known that disruption of the MMP/TIMP activity ratio
with a gain-of-function of proteasic activity over basement membrane and
extracellular matrix proteins is present in malignant tumors and parallels the
invasive potential (Tobar et al., 2010; Choi et al., 2011b). Then the key event
for demolishing the physiological tissue barrier (limits) and for invasion to
start is basically controlled by both HIF1α and NFkB through the expression of
these genes.
Adhesion molecules and counter-receptors
The activation of NFkB in leukocytes finely reprograms the expression of
adhesion molecules for migration and for homing at constitutive district tissue
or at damaged site. A NFkB-dependent and/or cytokine-dependent new expression of
adhesive molecules occurs also in tumor cells, allowing a number of biological
changes typically related with malignancy. These changes include the ability to
detach from the original tissue (i.e., cadherins), the ability to migrate
following a specific chemotactic gradient and a path of ECM molecules (receptors
for chemokines and integrins), and, finally, the identification of the homing
site represented by activated endothelial cells (ICAM-1, selectins, and their
countereceptors; Marcu et al., 2010).
Ongoing Clinical Trials and Future Directions
HIF1α
The recognition of the central role of HIF in tumor growth and progression and
the in vitro and in vivo demonstration of tumor growth inhibition by many HIF1α
inhibitors, is currently translating in clinical trials for some of them. In
particular, 2ME2 is undergoing Phase II clinical trials in patients with breast,
prostate, and ovarian cancer (Semenza, 2012; Xia et al., 2012). Similarly also
molecules derived from 2ME2 have been selected for evaluation of Phase I
clinical trials (Semenza, 2012; Xia et al., 2012). Analogs of geldanamycin (GA)
are in Phase II clinical trials in patients with VHL disease, breast cancer, etc
(Semenza, 2012; Xia et al., 2012). Bortezomib (velcade) has been approved in the
US by FDA for use in multiple myeloma, based on the results from the Phase II
trial. Two open-label, Phase III trials established the efficacy of bortezomib
1.3 mg/m2 (with or without dexamethasone) in patients with relapsed/refractory
multiple myeloma (Semenza, 2012; Xia et al., 2012). EZN-2968, is currently in
Phase I clinical trials (Semenza, 2012; Xia et al., 2012).
Finally, an important consideration is that many of HIF1α interacting drugs are
in clinical cancer trials or are already approved for the treatment of cancer or
other diseases (Xia et al., 2012).
NFkB
Several classes of NFkB inhibitors are currently being tested in conjunction
with chemotherapy and radiotherapy. In fact, a large number of clinical trials
are testing the efficacy and specificity of rationally designed drugs that
inhibit NFkB (for a specific trial in the US see the NIH service at:
ClincalTrials.gov). In the case of NFkB for example the proteasome inhibitor
Bortezomib (also used to inhibit HIF1α) has surprisingly limited side effects
and therefore currently appears to be no reason to reject NFκB as drug target on
the basis of potential adverse effects which might be induced by inhibition of
this transcription factor (Wilczynski et al., 2011). Recently, in a phase II
clinical trial, curcumin was found to be beneficial for patient with advanced
pancreatic cancer (Wilczynski et al., 2011; Grynkiewicz and Slifirski, 2012).
These are only two examples of a large number of NFkB inhibitors that are being
currently tested. Hopefully, in the next few years several NFkB inhibitors that
can increase the therapeutic efficiency of chemotherapy and radiotherapy will be
successfully employed in treatment of cancer patients.
Sirtuins
Considering the relatively short time that these enzymes are under
investigation, very few sirtuin modulators are under trials. In fact, currently,
there are no modulators that can specifically regulate a single sirtuin.
However, resveratrol and a number of its derivatives have shown beneficial
effects on a randomized double-blind cross-over trials in humans with effects
similar to calorie restriction and activation of AMPK, SIRT1, and PGC-1α levels
(Timmers et al., 2011). Inhibitors of SIRT1 and SIRT2 have been proposed for
treatment of cancer but are far from clinical trials (Morris, 2012).
Conclusion
Considering the central role of HIF1α and NFkB in metabolic reprogramming,
inflammation, and cancer, and considering also the fact that both transcription
factors are regulated by sirtuins, the possibility to develop more specific
modulators acting on different steps of these molecular net, hold promising
results toward new therapies with higher success rates.
Curcumin elevates sirtuin level but does not postpone in ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4991376
Apr 12, 2016 · Sirtuin 6 is able to deacetylate histone H3 at lysine 9 at the
promoter of RELA (component of NFκB), causing inhibition of transcription and
loss of NFκB activity . Taking into account that curcumin increased the level of
sirtuin 6, it can be assumed that there is …
Cited by: 14
Publish Year: 2016
A
Natural polyphenols as sirtuin 6 modulators | Scientific ...
https://www.nature.com/articles/s41598-018-22388-5
Mar 07, 2018 · Natural polyphenols as sirtuin 6 modulators. They have been shown
to modulate the activity of a NAD + -dependent histone deacetylase, SIRT6.
Because SIRT6 has been implicated in longevity, metabolism, DNA-repair, and
inflammatory response reduction, it is an interesting target in inflammatory and
metabolic diseases as well as in cancer.
Cited by: 22
Publish Year: 2018
Author: Minna Rahnasto-Rilla, Minna Rahnasto-Rilla, Jonna Tyni, Marjo Huovinen,
Elina Jarho, Tomasz Kulikowi...
Author: Minna Rahnasto-Rilla
Effects of Gallic Acid and Its Derivates on Inflammatory ... biomedpharmajournal.org/vol11no3/effects-of-gallic-acid-and-its-derivates-on... Gallic acid has 1 H group on the carbon side chains with ClogP value 0.89, while heptyl gallate with the chemical formula -(CH2)6-CH3 with ClogP value 2.32, and octyl gallate (CH2)7-CH3 with ClogP value 3.72 17 Heptyl and Octyl gallate is a synthetic derivative of gallic acid by adding an … Cited by: 1 Publish Year: 2018 Author: Arleni Bustami, Popi Sopiah, R. Muharam, Heri Wibowo Effect of Gallic acid in potentiating chemotherapeutic ...
https://cancerci.biomedcentral.com/articles/10.1186/s12935-019-0868-0
Jun 03, 2019 · The ability of Gallic acid to up regulate p53 seen in our study
is in agreement with Wang et al. who revealed that Gallic acid exhibited its
anticancer effect on human SCLC H446 cells via the ROS-dependent mitochondrial
apoptotic pathway. Cited by: 2 Publish Year: 2019 Author: Nora M. Aborehab, Nada
Osama Author: Nora M. Aborehab, Nada Osama Gallic Acid Regulates Body Weight and
Glucose Homeostasis ... https://academic.oup.com/endo/article/156/1/157/2800626
Jan 01, 2015 · Gallic acid [3,4,5-trihydroxybenzoic acid (GA)], a natural
phytochemical, is known to have a variety of cellular functions including
beneficial effects on metabolic syndromes. However, the molecular mechanism by
which GA exerts its beneficial effects is not known. Cited by: 54 Publish Year:
2015 Author: Khanh V. Doan, Chang Mann Ko, Ann W. Kinyua, Dong Joo Yang, Yun-Hee
Choi, In Young Oh, Nguyen Minh N... Author: Doan, Khanh V. Effect of Gallic Acid
on Dementia Type of Alzheimer ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4892325 Effect of Gallic Acid on
Dementia Type of Alzheimer Disease in Rats: Electrophysiological and
Histological Studies Somayeh Hajipour , 1 Alireza Sarkaki , 1 Yaghoob Farbood ,
1 Akram Eidi , 2 Pejman Mortazavi , 3 and Zohreh Valizadeh 4, *
Anti-inflammatory potential of ellagic acid, gallic acid ...
https://bmccomplementalternmed.biomedcentral.com/articles/10.1186/s12906-017-1555-0
Jan 14, 2017 · The inhibitory effect of ellagic acid, gallic acid and
punicalagin A&B were evaluated on the production of LPS-induced NO by Griess
reagent, PGE-2 and IL-6 by immunoassay kit and prostaglandin E2 competitive
ELISA kit, and COX-2 by Western blotting.
Cited by: 32
Publish Year: 2017
Author: Lamees A. BenSaad, Kah Hwi Kim, Chin Chew Quah, Wee Ric Kim, Mustafa
Shahimi
Author: Lamees A. Bensaad
肿瘤代谢中的缺氧信号通路:共同选择相互关联的生理通路的重要性
Hypoxia signaling pathways in cancer metabolism: the importance of co-selecting
interconnected physiological pathways
诺玛·马森 Norma Masson & 和彼得·拉特克利夫 Peter J Ratcliffe (note: cowinner of 2019
Noble Prize for medicine/phisiology)
牛津大学缺氧生物实验室
摘要
肿瘤缺氧和代谢紊乱都是癌症的典型特征。最近的分析显示,在癌症中,致癌激活、缺氧信号系统和代谢途径之间存在着复杂的相互联系。这些研究表明,代谢和缺氧信号通路不仅对能量消耗或肿瘤缺氧引起的错误信号作出简单的反应,而且在许多点上也与致癌信号机制直接相关。本文综述了目前对缺氧诱导因子(HIF)在这些网络中的作用的认识。它还将讨论这些相互关联的通路在产生癌症表型中的作用;特别是,从生理学上“硬连接”到导致癌症的致癌机制的大量通路的转换的影响。
评论
改变的能量代谢是癌症的经典特征,它在正电子发射断层扫描(FDG-PET)中支持标记的氟代脱氧葡萄糖的诊断用途,并且是定义该疾病新治疗方法的主要努力的重点。在正常细胞中,葡萄糖通过Embden-Meyerhof糖酵解途径转化为丙酮酸,然后主要注入线粒体中产生ATP。即使在有足够的氧气支持线粒体代谢的情况下,癌细胞也具有增强的“发酵”葡萄糖丙酮酸然后乳酸的能力。大约90年前,Warburg首次描述了这种作用,他坚信主要缺陷在于线粒体功能,著名地指出(癌症)“呼吸总是受干扰,因为它无法导致发酵消失”
[
1]。后来有争议的评论家对沃伯格为何如此坚定地坚持这一观点感到困惑,特别是因为他自己的氧气吸收数据并未明确支持这一观点[2]。沃伯格还与当前的思想保持一致,他还谈到糖酵解,据我们所知,糖酵解为生长提供了动力[3]。然而,很明显,不仅许多癌症中的线粒体能力与正常组织具有广泛的可比性,而且糖酵解的上调在癌症中并不普遍[2]。对实验性肿瘤的研究表明,尽管有氧糖酵解具有转移和杀死宿主的潜能,但在缓慢生长的肿瘤中好氧糖酵解并没有显着提高,但与分化差的快速生长变体密切相关。揭示出这种变化与基因表达向胎儿组织特有的特定酶同工型的转变显着相关[4,5]。
尽管有这些观察结果,但在20世纪后半叶,对肿瘤发生的遗传见解使对癌症新陈代谢的关注黯然失色,这涉及直接参与调节细胞周期,DNA修复,生长,凋亡和相关过程的分子。同样很清楚的是,Warburg所描述的代谢失调并不完全针对癌症,例如在免疫激活中的分裂细胞中观察到[6,7]。尽管这导致癌症研究重新聚焦于已从基因上确定的癌基因和肿瘤抑制途径,但人们对引起人们对癌症代谢途径的了解的兴趣重新兴起,其中包括负责的机制,这些变化支持肿瘤发展的方式以及这是否提供治疗途径。
除代谢改变外,肿瘤缺氧和缺氧信号通路的激活一直被确定为与侵袭性恶性肿瘤密切相关的特征。缺氧诱导因子(HIF)已被定义为介导对缺氧反应的关键转录因子,并且HIF目标基因与涉及肿瘤代谢失调的基因强烈重叠。这项审查将集中在HIF系统和癌症中的代谢变化之间的接口,以及激活复杂,相互联系,缺氧和代谢信号通路对癌症表型的影响。
HIF羟化酶系统
缺氧诱导因子-1(HIF-1)首先被鉴定为与促红细胞生成素基因的缺氧反应元件结合的转录调节因子[8]。最初认为,潜在的氧气传感设备仅限于促红细胞生成素的调节。然而,对缺氧反应元件的早期研究出乎意料地表明,该反应途径的作用范围更广[9]。随后清楚的是,HIF-1具有多种其他转录靶标,首先被鉴定的是编码多种糖酵解酶的基因[10,11]。有趣的是,这些研究表明,HIF的调节特异于特定的酶同工型,HIF的同工型特异的调节模式与先前在癌细胞中对Warburg效应的分析中所发现的惊人地相似,这引起了一个问题。关于HIF在癌症代谢改变中的作用[12]。
HIF系统本身已在其他地方进行了广泛的审查(有关更多详细的审查,请参见[13,14])。简而言之,HIF是α和β亚基的异二聚体,它们都是bHLH-PAS域(基本-Helix-Loop-Helix
Per-AHR / ARNT /
Sim)蛋白。存在三种HIF-α亚型(1α,2α和3α),其中对HIF-1α和HIF-2α的研究最为深入,并与HIF-1β形成转录活性异二聚体。低氧诱导的行为由HIF-α亚基赋予,其蛋白丰度和转录活性分别受氧依赖性脯氨酰和天冬酰胺基羟基化的调节(图1)。
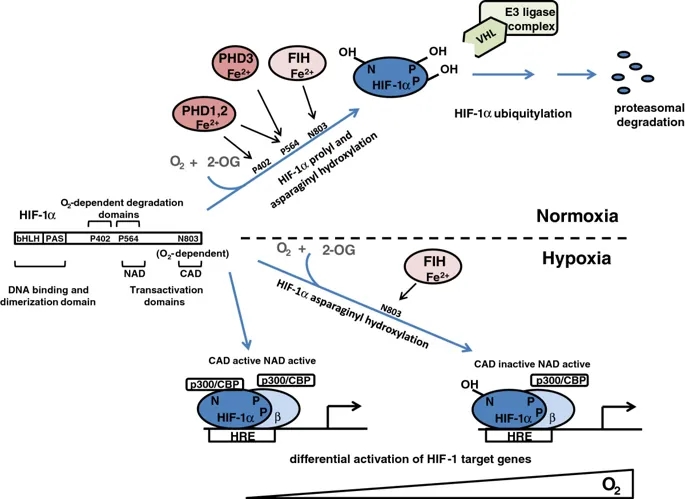
氧依赖性脯氨酰和HIF-1α的天冬酰胺基羟基化对HIF-1的调节。缺氧诱导因子(HIF)-1α是基本的Helix-Loop-Helix Per-AHR / ARNT / Sim(bHLH-PAS)域蛋白,包含三个残基,它们是调节性羟基化的靶标。 P402和P564被脯氨酰羟化酶结构域(PHD)酶靶向(请注意,PHD3只能使P564羟化),而N803通过抑制HIF(FIH)来靶向。 P402位于N端,P564位于C端O2依赖性降解域。脯氨酸羟化的HIF-1α被von Hippel-Lindau肿瘤抑制因子(pVHL)E3连接酶复合物识别,导致正常氧水平下降。有趣的是,脯氨酰和天冬酰胺基羟基化对缺氧有不同的敏感性。单独抑制脯氨酰羟基化(右下)足以使HIF-1α从依赖pVHL E3的蛋白水解破坏中逃脱,并通过N末端激活域(NAD)的活性与HIF-β形成活性转录复合物。在更严重的缺氧中,HIF-1α天冬酰胺基羟基化也被抑制(左下),从而允许将p300 / CBP共激活因子募集到其C末端反式激活域(CAD),并增强一组特定的HIF-1靶标的转录基因。 (HRE,缺氧反应元件)。
这些过程中最重要的是HIF脯氨酰羟基化,它对人类细胞中的HIF活性起着最重要的控制作用,并且在所有动物物种中都被保守。在高等动物中,HIF脯氨酰羟化发生在两个位点(人HIF-1α中的P402和P564)。在这些位点的羟化作用促进HIF-α多肽与von
Hippel-Lindau肿瘤抑制因子(pVHL)E3连接酶的β结构域缔合,导致泛素-蛋白酶体途径破坏HIF-α。尽管脯氨酰羟化的两个位点可以独立运行,但在天然HIF-1α和HIF-2α分子中,这些位点依序被羟基化并协同作用以增强pVHL介导的蛋白水解效率。在第二种调节途径中,天冬酰胺基羟基化(人HIF-1α中的N803)降低了C末端反式激活域(CAD)的活性,至少部分是通过阻止募集p300
/ CBP共激活因子来实现的(在[13 ])。
这些羟基化作用均由Fe(II)和2-氧代戊二酸酯(2-OG)依赖性双加氧酶超家族成员催化(有关综述,请参见[15,16])。作为双加氧酶,这些酶分解分子氧并将两个原子直接结合到它们的反应产物中。分子氧作为底物的绝对要求赋予了对缺氧的敏感性,尽管这可能会受到其他因素的调节。主要底物(HIF-α)的氧化与2-OG的氧化脱羧反应生成琥珀酸酯。偶联过程的失败会使酶处于非活性的氧化状态,因此需要Fe(II)催化中心的再生才能发挥活性。提出该方法是依赖于还原剂抗坏血酸盐的基础,该抗坏血酸盐是再生活性Fe(II)酶所必需的。催化铁的配位相对不稳定,因此酶是铁依赖性的,并且催化作用容易被铁螯合剂抑制。在人类细胞中,HIF脯氨酰羟化由三种密切相关的酶PHD(脯氨酰羟化酶域)1、2和3(也称为EGLN
2、1和3)催化,而HIF天冬酰胺基羟化则由一种酶FIH催化(抑制HIF的因素)。这些酶(2-OG,Fe(II)和抗坏血酸,以及分子氧)对多种共底物和辅因子的需求可能允许通过氧化还原,代谢和低氧刺激来调节HIF活性。在癌症中,所有这些信号都可能通过削弱一种或多种HIF羟化酶的活性来促进HIF通路的上调(图2)。
致癌信号通过多种平行途径激活HIF及其靶基因。除缺氧调节外,缺氧诱导因子(HIF)-α的羟基化作用还可能受代谢和氧化还原信号的影响。在癌症中,所有这些微环境应力都可以抑制羟基化,从而导致HIF的积累。致癌信号还会在许多其他方面影响HIF途径,包括转录,翻译,翻译后修饰和pVHL介导的HIF-α多肽降解。另外,致癌信号直接激活许多HIF靶基因。
此外,与氧感测过程不直接相关但通过对一个或多个HIF亚基的转录,翻译,翻译后修饰和蛋白质相互作用的影响来调节HIF途径的其他相互作用也是该研究的目标上调癌症中HIF的致癌过程(图2)。
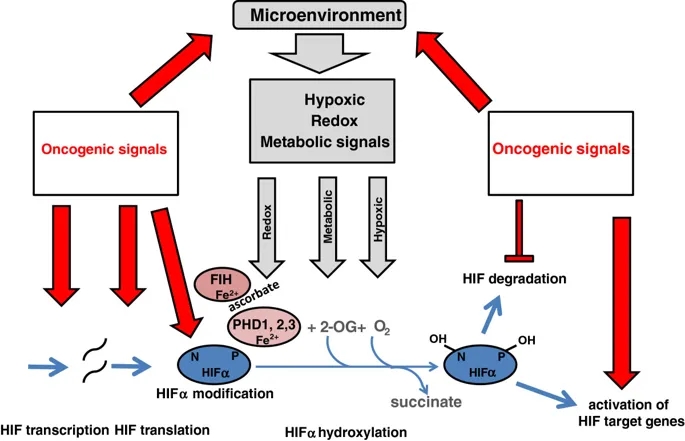
癌症中缺氧信号通路的上调
肿瘤缺氧
严重的缺氧和坏死区域在实体瘤中很常见,它们的存在与积极的临床进程相关。关于肿瘤坏死区域与血管分布有关的经典研究表明,肿瘤坏死至少部分是由缺氧引起的[17]。最近,这已被证明在邻近坏死区域的区域显着上调HIF靶基因[18]。在HIF遗传缺陷的实验性肿瘤中不存在这种基因表达模式,明确表明它是由这种途径驱动的[19]。
鉴于HIF靶基因包括许多支持肿瘤代谢失调的基因[20],并且肿瘤缺氧[21]和代谢失调的程度[2,5]与侵袭性癌症表型之间有着明显的相关性,因此很容易得出结论,即激活肿瘤缺氧本身引起的HIF通路异常是肿瘤代谢失调的主要原因。但是,许多观察表明,互连更为复杂。首先,在实体瘤中,缺氧区域(通过体内成像或使用组织化学标记进行评估)与HIF上调区域的同时发生率低于预期[22-24]。其次,不涉及实体组织肿块的血液系统恶性肿瘤也表现出HIF的上调[25,26]。总之,这些研究表明,尽管微环境缺氧明显促进了癌症中HIF的活化,但其他因素也必须很重要。
HIF途径的代谢调节
多种代谢途径影响HIF的调节,增加了以下可能性:除了HIF激活驱动癌症中代谢失调外,代谢失调还促进HIF激活。一种可能是2-OG可用性的改变会调节HIF羟基化。除其在克雷布斯循环中的作用外,2-OG还可作为谷氨酸脱氢酶还原酰胺化/氧化脱酰胺作用的共底物/产物,并且是转氨酶的主要氨基受体。因此,2-OG可以很好地用作调节HIF羟化酶活性的代谢传感器。与之相符的是,最近有报道称,在缺乏氨基酸的培养细胞中减少的细胞内2-OG可以降低PHD活性[27]。出乎意料的是,在这种情况下,PHD活性的调节对HIF-α蛋白水平没有影响,但是据报道会影响氨基酸激活mTORC1。尽管在这种情况下限制2-OG的利用是否能调节HIF的羟化还不清楚,但是PHD酶被认为是将氨基酸的利用与mTORC1途径联系起来的代谢传感器。
其他克雷布斯循环中间体和内源性有机酸代谢物也可能通过在催化位点(富马酸酯和琥珀酸酯)与2-OG竞争或通过产物抑制(琥珀酸酯)来改变HIF羟化酶的活性[28,29]。另外,在不同的测定中,据报道柠檬酸,异柠檬酸,苹果酸和草酰乙酸结合或抑制重组HIF羟化酶。不同的HIF羟化酶对这些抑制剂的敏感性不同。例如,富马酸盐比PHI更有效抑制PHD,而柠檬酸比PHD更有效抑制FIH
[28,30]。富马酸酯水合酶(FH,遗传性平滑肌瘤病和乳头状肾细胞癌)和琥珀酸脱氢酶(SDH,遗传性副神经节瘤)失活相关的遗传性癌症中,富马酸酯和琥珀酸酯均达到很高的水平[31-33]。在这些情况下,富马酸酯和琥珀酸酯至少部分通过抑制PHD酶而明显诱导出HIF
[30]。但是,在常见癌症中,Krebs循环和其他代谢中间体是否达到以及抑制HIF羟化酶所需的水平尚不清楚。
有趣的是,许多研究表明,在细胞培养基中提供葡萄糖和/或进行中的葡萄糖代谢对于缺氧诱导HIF-1α是必需的[34]。不同的研究者为一系列机制提供了证据。乳酸或丙酮酸的提供已显示可稳定葡萄糖缺乏的细胞培养物中的HIF-1α,并且已表明这种作用是由乳酸脱氢酶对乳酸产生的丙酮酸抑制PHD介导的[35]。乳酸通常在肿瘤中积累高水平(10
mM),并且还被提议激活癌症中的HIF和血管内皮生长因子(VEGF)介导的血管生成[36]。奇怪的是,在标准的(含抗坏血酸盐)反应条件下,丙酮酸和乳酸都不能与2-OG竞争或抑制纯化的重组PHD
[28,29]。最近的工作提出了一种不同的抑制方式,可以使人们对这一悖论有所了解,即丙酮酸和草酰乙酸通过氧化使PHDs失活,而抗坏血酸则可以逆转[37]。因此,代谢中间体具有通过至少两种机制抑制HIF羟化酶的潜力。与2-OG和氧化竞争。
最近,人们的兴趣也集中在另一种“竞争性代谢物”上,即2-羟基戊二酸酯(2-HG),它具有竞争性抑制2-OG双加氧酶的潜力。在低和中度神经胶质瘤,继发性胶质母细胞瘤,急性髓细胞性白血病中,高频率观察到了编码异柠檬酸脱氢酶(IDH)1和2的基因的特定突变,而在其他恶性肿瘤(包括骨髓增生异常综合征,T-细胞淋巴瘤,软骨肉瘤和胆管癌[38,39]。在受影响的细胞中,由于异常酶将2-OG还原而形成了2-HG,并积累到很高的水平[40]。然而,2-HG,特别是由突变IDH酶形成的“
R”对映异构体,对PHD酶的抑制作用较弱[41]。因此,不可能在这些情况下导致HIF的任何上调,甚至据报道会激活PHD2,导致HIF降低[42]。
除了代谢物对“氧感应”羟基化反应的影响外,在其他水平上还定义了代谢和HIF信号通路的多种相互作用。例如,HIF-α的水平受营养和细胞能量敏感的mTOR复合物的作用而受到复杂的翻译控制,HIF-1α受mTORC1和2的调节,而HIF-2α主要受mTORC2的调节[43]。在正常细胞中,存在几种机制,低氧可以通过mTOR途径或通过调节eIF2α或eEF2来减少翻译。这些途径本身独立于HIF,涉及AMPK或PERK的激活,以响应由缺氧引起的代谢变化[44]。但是,翻译控制也可以依赖于HIF。例如,HIF激活REDD1的转录[45],从而激活结节性硬化性TSC1
/ 2肿瘤抑制复合物[46],它是mTORC1的上游抑制剂。癌细胞可以至少部分通过mTOR复合物的致癌性失调规避HIF-α翻译的这种下调。
与新陈代谢的另一个界面是由HIF-α多肽特定位点的可逆乙酰化介导的。 HIF-1α可以被乙酰转移酶(例如p300 /
CBP相关因子(PCAF))在多个赖氨酸残基处乙酰化,乙酰化可以被几类酶逆转,包括经典的组蛋白脱乙酰酶(HDACs)和沉默调节蛋白[47-49]。由于它们对能量状态的另一个关键参数敏感,即细胞NAD
+ / NADH比率,瑟土因蛋白与HIF之间的界面引起了广泛的关注。赖氨酸脱乙酰基酶的哺乳动物sirtuin家族(SIRT1-SIRT7)将脱乙酰基与NAD +
+水解结合在一起。
SIRT1、3和6都与HIF活性的调节有关[48,50–53],尽管在互连的确切性质上存在分歧。在一项研究中,据报道,SIRT1使K674的HIF-1α脱乙酰化可阻止HIF-1α募集p300,而缺氧条件下SIRT1的失活(通过降低NAD
+水平)释放了该阴性对照[48]。另一项研究报告了缺氧条件下HIF-1α的全部活性需要SIRT1活性[53],而另一项研究则报道了SIRT1和HIF-2α之间的特定功能相互作用导致缺氧条件下HIF-2α反式激活的上调[52]。
]。在某些[54],但不是全部设置中,SIRT1本身也是HIF靶基因[53]。
铁,抗坏血酸和氧化应激
Fe(II)在HIF羟化酶催化中心的结合(如其他2-OG双加氧酶的结合)相对不稳定。这些酶还需要抗坏血酸来维持活性的Fe(II)催化中心。这些特性使它们易于受到氧化还原信号和铁的可利用性的调节,从而引发了关于氧化还原状态和/或铁的可利用性的异常是否在迅速分裂的癌细胞中异常代谢与HIF激活之间提供了另一个联系的问题。
在组织培养中,补充铁或抗坏血酸可促进HIF羟化酶活性并抑制含氧细胞中基础HIF的水平[55]。癌症患者通常营养不良,全身铁缺乏症很普遍[56]。此外,快速生长和/或血液供应不足可能加剧肿瘤中的细胞铁和抗坏血酸缺乏症。令人惊讶的是,尚未深入研究铁和抗坏血酸缺乏可能是导致临床癌症中HIF活化的重要因素的可能性。在抗坏血酸的啮齿动物中,补充抗坏血酸不会影响HIF激活的生理学指标,例如促红细胞生成素的产生,这表明组织培养研究可能并不代表完整生物体的作用[57]。然而,近来低细胞抗坏血酸与临床子宫内膜癌中HIF增加和侵袭性表型有关[58]。
实验研究强烈支持铁缺乏和氧化还原应激上调肿瘤中HIF的能力。例如,在乳腺癌细胞系异种移植模型中,已证明通过shRNA介导的转铁蛋白受体1敲低抑制铁摄取可激活HIF并增强血管生成[59]。在源自Ki-Ras转化的成纤维细胞的肿瘤中,据报道junD激活抗氧化反应可增强PHD活性,降低HIF并损害血管生成[60]。在另一个异种移植模型中,发现抗坏血酸和抗氧化剂N-乙酰半胱氨酸都通过增加羟化作用来抑制HIF激活和人类淋巴瘤细胞的生长[61]。对纯化的重组酶的动力学研究表明,除抗坏血酸盐以外的还原剂在激活HIF羟化酶中只能部分替代抗坏血酸盐[62],并且可能在细胞中,这些试剂间接作用于影响细胞Fe(II)的过程。
)或抗坏血酸水平。
也有人提出在缺氧状态下增加线粒体活性氧的产生会通过损害HIF羟化酶的活性来促进HIF的活化(在[63]中进行综述)。然而,是否应用线粒体抑制剂后HIF的激活减少是由于活性氧的减少,还是由于细胞内氧气水平的增加(由于线粒体耗氧量的减少)引起的(在文献[64]中进行了综述)。有趣的是,HIF天冬酰胺基羟基化比HIF脯氨酰羟基化对过氧化氢的抑制作用更为敏感[65],而对于缺氧抑制则相反[66]。这表明缺氧和活性氧通过不同的机制影响HIF信号传导。由于除最严重的缺氧水平外,HIF天冬酰胺基羟基化仍然持续[66],并特异性调节某些而非全部HIF靶基因的表达[67],这些发现也表明肿瘤中的缺氧和氧化还原信号之间的相互作用不仅激活了HIF,但影响了HIF转录反应的性质。
除了调节HIF羟化酶活性外,氧化还原信号和氧化应激(例如代谢失调)还可以许多其他水平影响HIF途径。在单个HIF-α亚型的转录和翻译中均观察到影响。例如,HIF-1α启动子包含一个特征明确的核因子κB(NF-kB)结合位点,可通过氧化应激传递上调[68],而HIF-1β转录也可被NF-kB直接激活[
69]。烟酰胺腺嘌呤二核苷酸磷酸(NADPH)氧化酶在pVHL缺陷型肾癌细胞系(其中pVHL依赖性蛋白水解被破坏)中的作用研究表明了其他几个控制水平。例如,NADPH氧化酶,特别是Nox4,被发现可以提高HIF-2αmRNA的水平[70],而NADPH氧化酶依赖性的活性氧的产生被认为可以增强HIF-2α的翻译[71]。综上所述,这些发现揭示了与氧化还原信号的多种相互作用,其可能影响癌症中HIF途径激活的定量和定性方面。
致癌和抑癌途径
除了通过多种微环境刺激激活外,HIF系统还通过多种肿瘤抑制因子和致癌基因途径激活。其中最引人注目的是von
Hippel-Lindau肿瘤(VHL)抑制子的突变(综述[72])。如上所述,pVHL是泛素E3连接酶复合物的一部分,该复合物将HIF-α亚基靶向泛素-蛋白酶体途径。
VHL的双等位基因失活因此阻止了HIF-α的氧依赖性蛋白水解,并导致HIF途径的组成性激活。然而,有趣的是,在与pVHL相关的癌症中对HIF进行更详细的分析表明,定量作用对HIF激活的重要性。特别是,在受VHL疾病影响的家庭中,特定突变对HIF失调的定量影响与不同类型的瘤形成的患病率之间存在明显的相关性[73,74]。
HIF的严重失调与肾癌的易感性有关,但似乎与pVHL相关的嗜铬细胞瘤不相容,后者与部分失活的突变相关,从而导致更适度的HIF途径激活水平。
HIF还被一系列通过PI3K / PTEN / AKT或RAS / RAF /
MAPK信号传导级联作用的生长因子激活(在[75]中进行了综述)。在许多类型的癌症中,通过体细胞突变和基因扩增激活这些途径很常见,而PI3K / PTEN /
AKT途径的失调会通过增加HIF-α亚基的合成而导致HIF的上调[76]。
AKT丝氨酸/苏氨酸激酶具有多个下游靶标,并可能通过mTOR依赖性和mTOR依赖性机制增加HIF-α的翻译[77]。
AKT也可能通过其他机制增加HIF-α水平。例如,AKT的另一种底物GSK3b已通过pVHL独立机制参与调控HIF-1α蛋白降解[78]。
据报道,RAS / RAF / MAPK途径主要通过反式激活的调控影响HIF活性。 HIF-1α或共激活因子p300被不同的激酶(p42 / p44
MAPK或p38)磷酸化可通过促进HIF / p300复合物的形成和增强p300反式激活来激活HIF [79]。
据报道,HIF和p53肿瘤抑制途径之间存在多种相互作用(综述见[80,81])。尽管并非所有报道都一致,但通常已证明诱导p53可抑制HIF活性。已经描述了p53和HIF-1α之间的直接物理相互作用和间接功能相互作用,包括p53和HIF-α之间对p300辅助激活剂的竞争[83],以及p53依赖于HIF-α促进HIF-α降解。小鼠双分钟2同源物(MDM2)泛素连接酶[84]。
基因突变
与经典的肿瘤抑制因子和与HIF系统相关的致癌途径中普遍存在的突变相反,HIF的直接突变激活(例如,通过降解域中关键残基的缺失或突变)在癌症中并不常见。相反,在pVHL缺陷型肾癌中,在HIF-1α中观察到少量但明显过量的失活突变,并且通过删除14q染色体区域减少HIF-1α基因的剂量是常见的[85-87]。在癌症中,HIF羟化酶的失活突变也不常见。
综上所述,这些发现揭示了多种手段和机制,通过这些手段和机制,微观环境和遗传改变均会导致癌症中HIF的上调。但是,他们还证明了HIF自身中与癌症相关的激活突变的低患病率与多种癌基因和肿瘤抑制基因(其产物会影响该通路的活性)中的突变的患病率之间存在显着差异。
据报道,RAS / RAF / MAPK途径主要通过反式激活的调控影响HIF活性。 HIF-1α或共激活因子p300被不同的激酶(p42 / p44
MAPK或p38)磷酸化可通过促进HIF / p300复合物的形成和增强p300反式激活来激活HIF [79]。
据报道,HIF和p53肿瘤抑制途径之间存在多种相互作用(综述见[80,81])。尽管并非所有报道都一致,但通常已证明诱导p53可抑制HIF活性。已经描述了p53和HIF-1α之间的直接物理相互作用和间接功能相互作用,包括p53和HIF-α之间对p300辅助激活剂的竞争[83],以及p53依赖于HIF-α促进HIF-α降解。小鼠双分钟2同源物(MDM2)泛素连接酶[84]。
基因突变
与经典的肿瘤抑制因子和与HIF系统相关的致癌途径中普遍存在的突变相反,HIF的直接突变激活(例如,通过降解域中关键残基的缺失或突变)在癌症中并不常见。相反,在pVHL缺陷型肾癌中,在HIF-1α中观察到少量但明显过量的失活突变,并且通过删除14q染色体区域减少HIF-1α基因的剂量是常见的[85-87]。在癌症中,HIF羟化酶的失活突变也不常见。
综上所述,这些发现揭示了多种手段和机制,通过这些手段和机制,微观环境和遗传改变均会导致癌症中HIF的上调。但是,他们还证明了HIF自身中与癌症相关的激活突变的低患病率与多种癌基因和肿瘤抑制基因(其产物会影响该通路的活性)中的突变的患病率之间存在显着差异。
另一个重要的调节酶是丙酮酸激酶。丙酮酸激酶催化糖酵解的最终步骤,因此可以很好地改变葡萄糖的代谢命运:或者最大化ATP的产生;或减慢糖酵解速度,导致提供生物合成途径的糖酵解中间体的积累。
HIF-1诱导PKM基因的转录[93]。然而,癌细胞用活性较低的替代剪接的胚胎形式(PKM2)代替了丙酮酸激酶(PKM1)的正常形式。转化为PKM2会改变主要亚基间接触域内的残基[94],并使PKM2活性更容易被可逆亚基从活性四聚体变为非活性二聚体的下调。改用PKM2还可促进上游酶磷酸甘油酸突变酶1(PGAM-1)的磷酸烯醇丙酮酸(PEP)依赖性组氨酸磷酸化的催化,从而增加PGAM-1的活性,并再次提出将糖酵解通量从ATP合成引向生物合成中间体的生产[95]。有趣的是,PKM2在支持癌症生长中的作用似乎与环境密切相关。确实,敲除PKM2在异种移植肿瘤模型中产生了矛盾的结果[96,97],最近对小鼠肿瘤发生的研究表明,虽然PKM2同工型特异性缺失可促进肿瘤生长,但它似乎也可以阻止组织培养细胞系的生长。肿瘤[98]。与初级转录本的上调相反,HIF是否参与将PKM1切换到PKM2的替代剪接过程尚不清楚。然而,有趣的是,最近的报告描述了PKM2与HIF系统的完全不同但特定的相互作用,描述了PKM2在HIF-1α的转录共激活中的非糖酵解作用,可能驱动正增强电路[93]。
结合糖酵解中间体在HIF调节中的拟议作用,这些发现表明HIF途径与糖酵解之间存在极其复杂的相互作用。但是,相对于酶和调节剂的水平,相对较少的研究实际测量了HIF依赖的代谢通量变化。转化的小鼠胚胎成纤维细胞的研究表明,在低氧培养中,HIF-1α的失活与乳酸生成减少和ATP维持水平降低有关[99]。因此,氧气对HIF的下调似乎至少部分地有助于哺乳动物细胞的“巴斯德效应”(在氧气存在下发酵的下调)。然而,有趣的是,这些研究并未揭示HIF失活对常氧细胞中乳酸产生的影响。同样,最近对Hct116结肠癌细胞的代谢谱分析表明,尽管siRNA介导的HIF-1β敲低可降低乳酸生成的低氧上调,但它并不会降低常氧或低氧细胞的葡萄糖摄取[100]。此外,对HIF-1β缺陷型肝癌细胞的研究表明,尽管在没有HIF的情况下多个糖酵解基因下调,但仍可通过FDG-PET分析葡萄糖摄取和通过测量乳酸输出来评估糖酵解通量。进一步的分析表明,通过增加HIF-1β缺陷细胞中AMP
/ ATP比率,增强的糖酵解通量与PFK-1的变构活化有关[101]。
因此,尽管癌细胞中HIF的激活以及HIF对特定糖酵解酶,调节剂和转运蛋白的作用都表明HIF对Warburg效应做出了重要贡献,但干预研究表明,至少在下半年,糖酵解的上调仍在继续。没有HIF的某些设置。对肿瘤抑制因子和致癌基因作用的研究揭示了与糖酵解调控和致癌糖酵解激活的平行途径有关的多个直接界面,大概解释了这些观察结果。尽管如此,HIF对糖酵解途径中几乎每个点的同功酶特异性靶向都强烈暗示,尽管通过平行途径,HIF还是有助于癌症中糖酵解的上调。
结合糖酵解中间体在HIF调节中的拟议作用,这些发现表明HIF途径与糖酵解之间存在极其复杂的相互作用。但是,相对于酶和调节剂的水平,相对较少的研究实际测量了HIF依赖的代谢通量变化。转化的小鼠胚胎成纤维细胞的研究表明,在低氧培养中,HIF-1α的失活与乳酸生成减少和ATP维持水平降低有关[99]。因此,氧气对HIF的下调似乎至少部分地有助于哺乳动物细胞的“巴斯德效应”(在氧气存在下发酵的下调)。然而,有趣的是,这些研究并未揭示HIF失活对常氧细胞中乳酸产生的影响。同样,最近对Hct116结肠癌细胞的代谢谱分析表明,尽管siRNA介导的HIF-1β敲低可降低乳酸生成的低氧上调,但它并不会降低常氧或低氧细胞的葡萄糖摄取[100]。此外,对HIF-1β缺陷型肝癌细胞的研究表明,尽管在没有HIF的情况下多个糖酵解基因下调,但仍可通过FDG-PET分析葡萄糖摄取和通过测量乳酸输出来评估糖酵解通量。进一步的分析表明,通过增加HIF-1β缺陷细胞中AMP
/ ATP比率,增强的糖酵解通量与PFK-1的变构活化有关[101]。
因此,尽管癌细胞中HIF的激活以及HIF对特定糖酵解酶,调节剂和转运蛋白的作用都表明HIF对Warburg效应做出了重要贡献,但干预研究表明,至少在下半年,糖酵解的上调仍在继续。没有HIF的某些设置。对肿瘤抑制因子和致癌基因作用的研究揭示了与糖酵解调控和致癌糖酵解激活的平行途径有关的多个直接界面,大概解释了这些观察结果。尽管如此,HIF对糖酵解途径中几乎每个点的同功酶特异性靶向都强烈暗示,尽管通过平行途径,HIF还是有助于癌症中糖酵解的上调。
脂质代谢
脂质合成异常是另一种与缺氧和HIF相互作用的生物合成癌症表型。脂质含量的增加是癌细胞的共同特征,在某些肿瘤中,大量积累有助于形成透明细胞表型。与糖原积累一样,如此大的储备积累的原因尚不清楚,尽管这两种现象似乎是相关的。例如,脂质和糖原均在透明细胞肾癌细胞中蓄积,其中缺陷性pVHL导致HIF的组成型激活。在正常培养基中失去了透明细胞表型,而在成脂培养基中的生长促进了糖原和脂质的积累[106]。
尽管能源成本高昂(16碳脂肪酸棕榈酸酯的每个分子14 ATP和7
NADPH),但脂质在癌症中的蓄积显然涉及从头脂肪酸合成的增加[107]。在该过程中,首先通过乙酰辅酶A羧化酶将细胞质乙酰辅酶A转化为丙二酰辅酶A。然后,通过在脂肪酸合酶(FAS)的催化下向碳链中连续添加丙二酰辅酶A来生产脂肪酸。
FAS的上调与侵袭性恶性肿瘤密切相关,FAS的抑制迅速抑制癌细胞的增殖,诱导细胞周期停滞和凋亡[108,109](综述参见[110])。脂质合成潜在地提供了用于生产对于细胞增殖重要的新膜和脂质信号分子的资源。据此,据报道在癌症中上调了非常广泛的脂质分子的合成,包括不同类型的磷脂,胆固醇和脂质激素,前列腺素,白三烯和鞘脂以及脂肪酸。
值得注意的是,低氧似乎促进了这些合成途径中的许多,并促进了细胞脂质的摄取,脂质和低氧信号传导途径之间的相互作用发生在多个水平[111,112]。通过大规模途径分析,对脂质合成基因表达模式的多重影响是显而易见的。例如,在pVHL缺陷型癌症中存在广泛的脂肪基因表达特征,其中HIF被组成性激活[106]。脂质代谢中的关键单个基因也据报道被缺氧强烈诱导。例如,在低氧组织培养中都诱导了胞浆形式的乙酰辅酶A合成酶(在胞质溶胶中生成用于脂肪酸合成的乙酰辅酶A)和FAS本身,而在肿瘤中,FAS表达的空间模式与低氧标志物一致[113]。
]。其他研究已经确定了与脂质生物学高度不同的过程有关的缺氧诱导基因。例子包括在脂质滴形成(缺氧诱导蛋白2),前列腺素生物合成(环加氧酶2),脂质信号系统(脂加氧酶12-lox,鞘氨醇激酶,SphK1)和合成过程(硬脂酰-CoA去饱和酶-1,单不饱和脂肪酸生物合成中的限速酶)[114-117]。
尽管这些基因中的一些是直接的HIF转录靶标,但在其他基因中,上调是通过HIF对调节脂质代谢或脂肪分化程序的转录因子的作用间接介导的。因此,据报道,在不同的环境中,HIF-1会下调过氧化物酶体增殖物激活受体(PPAR)-α,并上调或下调PPAR-γ[118-120]。缺氧还与许多其他转录网络相互作用,包括作用于脂质代谢关键靶点的固醇反应元件结合蛋白(SREBPs),DEC1
/ 2和GATA2 / 3。
例如,FAS的低氧诱导似乎涉及HIF依赖性SREBP1的上调和SREBP1对FAS启动子的作用[113]。
缺氧对提供脂质合成中间体的途径的影响也可能导致脂质通量发生变化(图3)。因此,至少在某些低氧环境下,丙酮酸脱氢酶(见下文)减少的乙酰辅酶A产生会损害葡萄糖的脂肪酸合成。通常,由丙酮酸脱氢酶在线粒体中产生的乙酰辅酶A被克雷布斯循环酶柠檬酸合酶转化为柠檬酸。当过量时,柠檬酸盐然后可以被转运到细胞质中,并通过ATP柠檬酸盐裂合酶转化回乙酰辅酶A。然后,将胞质乙酰辅酶A用于脂质生物合成。但是,在缺氧癌细胞中,葡萄糖代谢产物通过该途径的流动减少,谷氨酰胺的代谢可以补偿以维持细胞质乙酰CoA的供应。代谢通量研究表明,这是通过线粒体酶异柠檬酸脱氢酶2(IDH2)沿与Krebs循环相反的方向进行而发生的,该循环通过来自谷氨酰胺的2-OG的还原羧化作用生成柠檬酸[121]。还原性羧化的另一种胞质途径是由IDH1介导的[122,123]。这些途径在ATP-柠檬酸裂解酶作用下提供了细胞质乙酰辅酶A,代谢通量分析已证明,谷氨酰胺驱动的还原性羧化作用因缺氧而增加,并依赖于HIF
[122]。究竟HIF如何刺激还原性羧化尚不清楚,但是在pVHL缺陷型肾癌细胞中对HIF-α敲低的研究表明对HIF-1α和/或HIF-2α的依赖性,[121,122]导致细胞内柠檬酸盐的减少水平[124]。
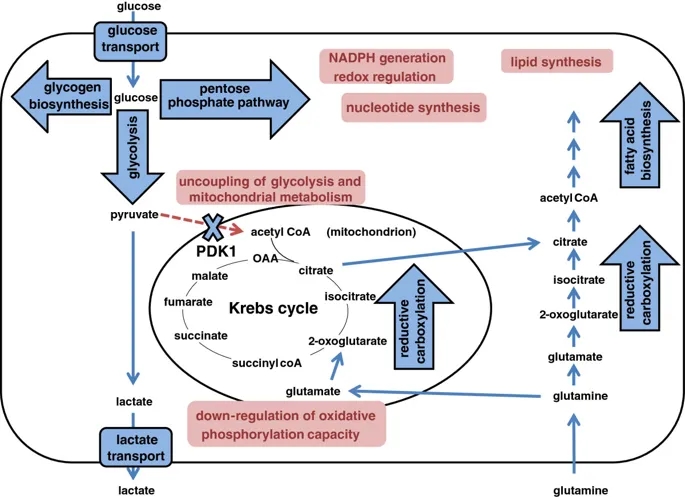
图示HIF在细胞代谢多个方面的作用。Schematic illustrating the action of HIF on multiple aspects of cellular metabolism.
线粒体代谢
在反映其在氧稳态中作用的过程中,HIF通过对线粒体代谢和生物发生的一系列作用来下调线粒体的氧化磷酸化。
在氧气存在下,由糖酵解产生的丙酮酸通过丙酮酸脱氢酶复合物(PDH)在线粒体基质中转化为乙酰CoA,为克雷布斯循环提供了底物。
PDH的活性受丙酮酸脱氢酶激酶(PDK)异构体1至4和丙酮酸脱氢酶磷酸酶(PDP)异构体1和2催化的磷酸化作用的调节,它们分别使其失活和活化。
HIF-1依赖的PDK1诱导导致PDH抑制,从而使Krebs循环与糖酵解断开(图3)[125,126]。
线粒体功能还可以通过依赖HIF的电子传输链中几个成分的活性下调来减弱。例如,据报道,复合物1的活性由于NADH脱氢酶[泛醌]
1α亚复合物4样2,NDUFA4L2的HIF-1依赖性活化而受到抑制[127]。复合物2,琥珀酸脱氢酶(SDH)复合物(SDHA,B,C和D)在缺氧时也表现出降低的活性。确切的机制尚不清楚,但据报道涉及通过转录后机制使HHB-1依赖性的SDHB蛋白水平降低[128]。电子传输链中的最后一种酶,细胞色素C氧化酶(COX),也是HIF依赖性调节的靶标。它的两个调节亚基通过HIF表现出不同的调节作用,从而允许切换到更适合低氧的亚基组成:COX4-2是HIF转录的靶标,在低氧条件下被上调;而COX4-1亚基则是通过(至少部分地)通过HIF依赖的降解COX4-1的线粒体LON蛋白酶的活化而间接调节的(至少部分)[129]。
HIF对线粒体功能和电子传输链的依赖效应也可以通过间接机制来实现。例如,HIF对microRNA
miR-210的转录激活导致对线粒体功能重要的多个靶标的下调,包括NDUFA4,SDHD,铁硫簇装配蛋白ISCU1 / 2和COX装配蛋白COX10
[130,131]。
最后,HIF可以通过影响整个细胞器水平来影响线粒体功能。对pVHL缺陷型肾癌细胞RCC4的研究表明,HIF可通过负调节c-MYC抑制线粒体呼吸和线粒体质量,其活性可促进线粒体的生物发生[132]。
HIF和MYC之间的串扰已定义为多个水平,包括在DNA结合位点的合作和竞争,特定HIF-α亚型和MYC亚基之间的非DNA结合相互作用以及由HIF-α亚型表达介导的相互作用。
HIF靶基因MXI-1,其抑制c-MYC活性[132]。另外,HIF靶基因BNIP3通过增加线粒体自噬而有助于减少线粒体数目[133]。
总体而言,HIF的这些作用会导致线粒体呼吸的适应性转变。通过还原性羧化反应生成柠檬酸盐,并保留生物合成能力,在缺氧条件下具有生存优势。
HIF-1α缺陷细胞在缺氧时不能下调氧气消耗,并且在缺氧状态下生存受损,至少部分原因是由于活性氧的产生增加[129]。
结论
总体而言,这些研究定义了多种途径,可通过多种手段上调HIF在癌症中的作用(图2)和HIF活化对癌症代谢的多种作用(图3)。此外,尽管HIF直接或间接作用于编码多种酶的基因,这些酶促成癌症的代谢失调,但许多这些酶还是致癌途径的直接靶标。例如,PI3K
/ PTEN /
AKT信号转导的激活会激活HIF,因此间接激活编码多种糖酵解酶的基因的转录。然而,AKT激活还可以通过一系列其他机制直接上调葡萄糖代谢,包括增加表达[134]和将葡萄糖转运蛋白运输到细胞膜[135,136],增加糖酵解酶的表达和改变调节磷酸化组件,例如PFKFB2
[137–139]。这些复杂路径的并行操作使得任何一种分子连接的非冗余作用都难以预测。
毫不奇怪,由于HIF主要是转录因子,因此大多数研究都集中在对基因表达的影响上,而关于HIF对特定代谢通量的影响的研究相对较少。但是,关键的酶活性在基因表达以外的许多水平上受到调节,并且新陈代谢的改变要求许多酶功能的协调变化。因此,很难从基因表达水平的测量中得出对代谢通量的总体影响,并且通过不同的代谢途径对HIF激活对通量的影响进行更直接的评估将是有用的。尽管有这些警告,但很明显,HIF会导致癌细胞代谢异常的许多方面。考虑到这些作用的多样性,一个重要但尚未完全解决的问题涉及HIF激活和致癌激活在同一代谢途径上同时发生作用的程度。
HIF激活的作用与致癌途径的作用之间最明显的区别是对线粒体功能的作用。
HIF激活下调线粒体的生物发生和线粒体的代谢,而致癌途径通常具有相反的作用,通过多种机制促进线粒体的代谢能力(综述[140,141])。由于线粒体代谢是细胞耗氧量的主要组成部分,HIF降低缺氧时的耗氧量的这种作用可以通过HIF在维持氧稳态中的核心作用来合理化,并且有人提出,在这种情况下HIF可以根据氧气的利用情况来调节癌症中的代谢异常。
与对线粒体代谢的影响相反,其他各种代谢功能在HIF激活的影响和激活的致癌途径之间也表现出相似的相似性。由HIF上调的糖酵解酶同工型与直接由致癌途径靶向的糖酵解酶同工型之间的一致性是惊人的。同样引人注目的是通过HIF和激活的致癌途径激活涉及过量糖原和脂质生产的生物合成途径之间的一致性。缺氧细胞中糖酵解增加的ATP产生显然与维持氧稳态有关。但是,由HIF和致癌途径两者诱导的糖酵解酶同工型激活方式并未最大限度地提高ATP的产生,大量证据表明其主要功能是生物合成中间体的供应。同样,糖原合成和脂质生物合成的激活(同样通过HIF和致癌途径)被认为反映了增殖细胞对生物合成中间体的需求(综述[110,142])。由于这些过程既不会减少氧气需求,也不会增加氧气供应,因此这些作用与HIF作为氧气稳态调节剂的概念之间的关系还不清楚。因此,可能有必要修改对HIF系统主要功能的看法,以包括对生长/修复途径(也许是对缺氧组织损伤的反应)的一般支持,而不是诸如血管生成或红细胞生成等更受限制的生长途径。直接增强氧气的输送。
不管有何解释,大量证据表明HIF参与支持癌细胞生长的生物合成途径的上调,而多种遗传和微环境改变则促进了HIF在癌症中的上调。鉴于HIF激活,侵袭性表型和不同类型癌症的不良预后之间存在令人信服的关联,因此很容易得出结论,HIF激活不仅是癌症表型的主要贡献者,而且与癌症的进展有因果关系。出人意料的是,尽管机械分析强有力地支持了这些陈述中的第一个,但仍缺乏人类遗传学证据来支持第二个陈述。
如上所述,癌症基因组分析尚未在编码任何HIF亚基的基因中定义典型的致癌激活突变簇。迄今为止,癌症中HIF突变选择的最明确证据是在pVHL缺陷型肾癌的背景下,HIF-1α的失活突变过多(综述[143])。
“共选”假设
我们先前已提请注意这一悖论,并认为HIF激活与侵略性癌症之间的异常强关联很可能反映出存在“硬连线”途径,这些途径在生理上起作用以将组织的生长与氧气供应联系起来,因此是“被直接驱动细胞增殖的致癌途径“选择”
[144]。这种“共选”假设的重要含义是,HIF转录级联的非常广泛的连接都将被“共选”为癌症表型(通过HIF的致癌或微环境激活),无论是否他们正在促进或限制癌症的发展。尽管HIF激活的许多方面都可能促进癌症的发展,但构成对低氧的生理反应的一部分的细胞生长抑制反应的存在可能具有相反的作用,这也许可以解释为观察到HIF-1α显示出某些肿瘤抑制因子的特征,至少在pVHL缺陷型肾癌的情况下[87]。因此,大量“硬连接”
HIF途径“共同选择”进入癌症表型可能会招致巨大的健康损失,并且与表型紧密相关的预后关联并不一定暗示因果关系。在考虑癌症中HIF激活与代谢失调之间的关系时,一个重要的问题是,类似的论点延伸到支撑癌症代谢失调的大规模复杂的HIF依赖性和HIF依赖性途径吗?
理解癌症代谢的一个关键进展是认识到失调的代谢途径不仅是对错误信号的响应,例如肿瘤团块内产生的代谢产物耗竭,而且(与HIF一样)还直接与致癌信号途径相关[
145,146]。此外,像HIF一样,存在类似的遗传悖论。尽管已经描述了许多在癌症中过度表达或激活的代谢酶的基因扩增事件[146],但对于大多数其上调与侵袭性癌症显着相关的代谢途径而言,明显缺乏突变直接激活通路。有趣的是,在特定的罕见癌症综合征中失活的非常特定的“代谢”抑癌基因(即FH和SDH)中确实存在着惊人的突变谱,这一悖论已成为人们更加关注的焦点[30-32],以及IDH1和2个癌基因。
IDH1和2的突变特征特别有用。在每个基因的高度特异性底物结合残基处观察到多个不同的取代,但在其他位置未观察到。这些突变是杂合的,符合经典的癌基因预期(主要作用是促进癌症),并且已经通过产生2-HG为其致癌作用收集了有力证据[40]。
2-HG对参与表观遗传调控或其他潜在致癌途径的2-OG依赖性双加氧酶的拟议作用的证据已在其他地方进行了综述[147]。对于当前的讨论而言,重要的是IDH1 /
2突变谱可作为引人注目的阳性对照,强调了人类癌症“纯化”确实具有选择性优势的突变的非凡能力。在这种背景下,属于癌症中上调的其他代谢途径的酶相对缺乏激活突变的现象令人震惊。与HIF一样,这与代谢酶和致癌基因突变之间的众多联系形成鲜明对比。
因此,似乎存在生理上支持分裂细胞的“硬连线”代谢途径的共选择可能因此扩展到了HIF依赖性和HIF依赖性途径,从而支持了癌症中代谢失调(图4)。尽管多项干预研究支持了代谢失调对癌症生长的积极贡献,因此,这些途径可能代表了抗癌治疗的潜在目标[105,148],但“共选”假说暗示了这一点无法推断。直接来自与侵袭性恶性肿瘤的密切联系,因此需要在特定癌症的情况下直接进行检测。
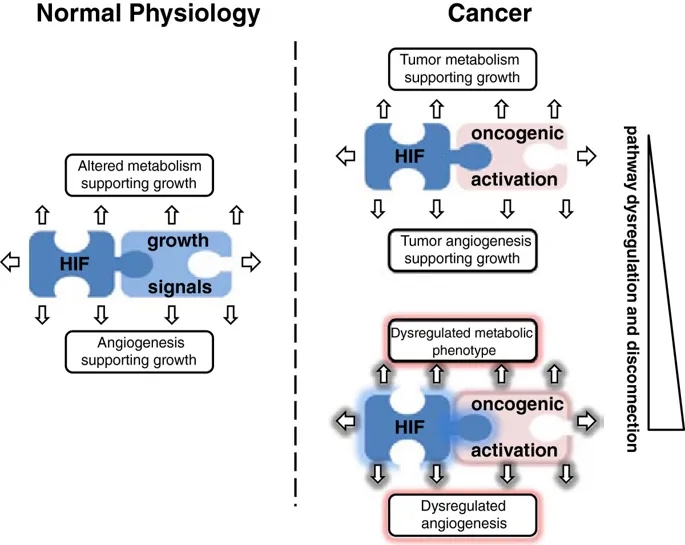
图4
示意图说明了促进生长的信号,HIF和代谢/血管生成途径之间的“硬连线”互连。在生理生长过程中,这些途径受到严格调节(左图)。在肿瘤发生中,生长的增加激活了“硬连线”连接,从而产生了癌症的代谢和血管生成特征(右上图)。由于不同的致癌信号与缺氧诱导因子(HIF)和代谢/血管生成途径在数量和质量上都有不同的联系,因此癌症中由体细胞突变引起的致癌信号的随机调节失调有可能``共同选择''严重紊乱的代谢和血管生成表型(右下面板)。
在这一论点的扩展中,共选可能解释了癌症代谢失调的一个令人费解的方面,即偶尔由于明显的能量存储积累而产生奇异的细胞表型。如上所述,糖原和脂质生物合成的增加是癌细胞的共同特征。虽然可以通过提供细胞生长所需的生物合成中间体来合理化这些过程的激活,但要理解某些癌症中与“透明细胞”表型相关的糖原和脂质的大量积累如何协助癌症生长要困难得多。类似的论点适用于癌症相关的血管生成。通过有效的血管生成来夹带氧气供应对于癌症的生长显然很重要,但是通常在肿瘤中观察到的奇怪而无效的过度血管生成活性更难以理解,尤其是在直接选择血管生成上调的假设下。我们建议这两者都可以代表“共同选择”的电路的作用,这些电路“被硬连接”至致癌基因激活,以支持生长组织的需求。在生理条件下,这些途径受到严格调节,因此增强的“代谢”和“血管生成”活性与细胞生长良好协调。在癌症中,这些过程通常也被很好地协调,以携带广泛的功能性代谢和血管生成支持途径。但是,对导致癌症中HIF激活的机制和途径的综述揭示了在质量和数量上各不相同的机制。
因此,通过基因突变通过细胞自主优势赋予个体致癌途径的随机激活,也有可能产生新陈代谢和血管生成途径的杂乱无章的激活,这取决于受影响的致癌基因与代谢之间预先存在的联系。
/血管生成途径。例如,在某些情况下,途径激活可能不足以支持癌症的生长(例如,血管生成不良的肿瘤),而在其他情况下(例如,糖原和脂质的大量储存以及与HIF的强而直接的激活相关的夸大的血管生成)
pVHL缺陷的肿瘤),它是过量的(图4)。
总而言之,有关HIF途径的研究揭示了癌症生长与HIF激活之间以及HIF激活与癌症表型之间非常复杂的交互联系。我们认为,这些发现最重要的,也是迄今为止未被充分理解的含义之一是,有必要考虑随着癌症的进展共同选择大量的硬连线生理途径的含义。尽管我们从HIF的角度回顾了这一考虑因素(在这种情况下,细胞容易暴露于低氧的生理刺激下已迅速认识到该途径的巨大复杂性),但类似的考虑因素也可能适用于其他因素。微环境压力或癌症的基因突变可能会激活这些通路。
Hypoxia signaling pathways in cancer metabolism: the importance of co-selecting interconnected physiological pathways
Norma Masson & Peter J Ratcliffe
The Hypoxia Biology Laboratory, The University of Oxford
Abstract
Both tumor hypoxia and dysregulated metabolism are classical features of cancer. Recent analyses have revealed complex interconnections between oncogenic activation, hypoxia signaling systems and metabolic pathways that are dysregulated in cancer. These studies have demonstrated that rather than responding simply to error signals arising from energy depletion or tumor hypoxia, metabolic and hypoxia signaling pathways are also directly connected to oncogenic signaling mechanisms at many points. This review will summarize current understanding of the role of hypoxia inducible factor (HIF) in these networks. It will also discuss the role of these interconnected pathways in generating the cancer phenotype; in particular, the implications of switching massive pathways that are physiologically 'hard-wired’ to oncogenic mechanisms driving cancer.
Hypoxia signaling pathways in cancer metabolism: the importance of
co-selecting interconnected physiological pathways
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3938304/
L-抗坏血酸:HIF脯氨酰羟化酶的真正底物吗?
L-ascorbic acid: A true substrate for HIF prolyl hydroxylase?
a
Dmitry Rogachev National Medical Research Center of Pediatric Hematology,
Oncology and Immunology, 117997, Moscow, Russian Federation
b
Department of Chemical Enzymology, Faculty of Chemistry, M.V. Lomonosov Moscow
State University, Moscow, 119992, Russian Federation
c
Department of Pharmacology and Toxicology, Medical College of Georgia, Augusta
University, Augusta, GA 30912, USA
d
Department of Medicinal Chemistry and Pharmacognosy, University of Illinois at
Chicago, 833 South Wood Street, Chicago, IL 60612, USA
e
Department of Anatomy and Cell Biology, New York Medical College, 15 Dana Road,
Valhalla, NY 10595, USA
f
Bach Institute of Biochemistry, Research Center of Biotechnology of the Russian
Academy of Sciences. 33, bld. 2 Leninsky Ave., Moscow 119071, Russian Federation
g
Innovations and High Technologies MSU Ltd, Tsymlyanskaya, 16, of 96, Moscow,
109599, Russian Federation
强调
•
L-抗坏血酸而不是D-异抗坏血酸或N-乙酰半胱氨酸,抑制HIF1 ODD-luc报告基因激活。
•
L-抗坏血酸的抑制取决于HIF脯氨酰羟化酶抑制剂的化学性质。
•
L-抗坏血酸的抑制显示出饱和行为,表明了Michaelis-Menten型相互作用。
•
L-抗坏血酸通过支链氧喹啉抑制剂抑制HIF靶基因VEGF和HO-1的诱导。
抽象
L-抗坏血酸盐(L-Asc),但不抑制D-异抗坏血酸盐(D-Asc)和N-乙酰半胱氨酸(NAC)抑制由各种HIF脯氨酰羟化酶(PHD)抑制剂诱导的HIF1
ODD-luc报告基因激活。 L-Asc抑制的效率对选择用于报道分子激活的HIF
PHD抑制剂的性质敏感。特别是,与α-酮戊二酸(αKG)竞争的抑制剂,对L-Asc(40-100μM)生理范围的抑制作用不如具有强铁螯合基序的抑制剂敏感。用D-Asc对报告系统中的HIF活化剂进行挑战表明,尽管D-异构体对铁的还原力相同,但与L-Asc相比几乎没有作用。同样,使用细胞渗透性还原剂NAC直至1
mM,也未观察到对报告基因激活的影响。 L-Asc和D-Asc酸对接到HIF
PHD2晶体结构中显示出Tyr310对D-Asc的干扰。这表明L-Asc不仅是防止酶失活的还原剂。相反,总体结果表明,L-Asc是HIF
PHD的共底物,可竞争酶活性中心中αKG的结合位点。该结论与最近在基于细胞的TET酶和jumonji组蛋白脱甲基酶的系统中获得的结果一致,其中L-Asc被提议作为共底物而不是作为防止酶失活的还原剂。
关键词
催化周期HIF1 ODD-萤光素酶报告基因检测适应症HIF PHD抑制剂TET酶Jumonji脱甲基酶
Highlights
•
L-Ascorbate, but not D-isoascorbate, or N-acetyl cysteine suppresses HIF1
ODD-luc reporter activation.
•
The suppression by L-ascorbate depends on the chemical nature of HIF prolyl
hydroxylase inhibitor.
•
The suppression by L-ascorbate shows a saturation behavior, indicative of the
Michaelis-Menten type interaction.
•
L-Ascorbate suppresses the induction of HIF target genes VEGF and HO-1 with the
branched oxyquinoline inhibitor.
Abstract
L-Ascorbate (L-Asc), but not D-isoascorbate (D-Asc) and N-acetylcysteine (NAC)
suppress HIF1 ODD-luc reporter activation induced by various inhibitors of HIF
prolyl hydroxylase (PHD). The efficiency of suppression by L-Asc was sensitive
to the nature of HIF PHD inhibitor chosen for reporter activation. In
particular, the inhibitors developed to compete with alpha-ketoglutarate (αKG),
were less sensitive to suppression by the physiological range of L-Asc
(40–100 μM) than those having a strong iron chelation motif. Challenging those
HIF activators in the reporter system with D-Asc demonstrated that the D-isomer,
despite exhibiting the same reducing potency with respect to ferric iron, had
almost no effect compared to L-Asc. Similarly, no effect on reporter activation
was observed with cell-permeable reducing agent NAC up to 1 mM. Docking of L-Asc
and D-Asc acid into the HIF PHD2 crystal structure showed interference of Tyr310
with respect to D-Asc. This suggests that L-Asc is not merely a reducing agent
preventing enzyme inactivation. Rather, the overall results identify L-Asc as a
co-substrate of HIF PHD that may compete for the binding site of αKG in the
enzyme active center. This conclusion is in agreement with the results obtained
recently in cell-based systems for TET enzymes and jumonji histone demethylases,
where L-Asc has been proposed to act as a co-substrate and not as a reducing
agent preventing enzyme inactivation.
L-ascorbic acid: A true substrate for HIF prolyl hydroxylase?
甲状腺病变中缺氧诱导因子1α和2α的表达及其与维生素C水平的关系
Expression of hypoxia inducible factor 1α and 2α and its association with
vitamin C level in thyroid lesions
摘要
背景
细胞通过转录诱导参与血管生成,葡萄糖代谢和细胞增殖调节的基因来适应缺氧。介导细胞对低氧张力的反应的主要因素是缺氧诱导因子(HIFs),氧依赖性转录激活因子。
HIF的α亚基的稳定性和活性受需要抗坏血酸作为辅助因子的羟基化反应控制。因此,细胞内维生素C的缺乏可能会导致HIFs过度活化。在这项研究中,我们调查了人类甲状腺病变中的维生素C含量是否与HIF-1α和HIF-2α蛋白水平相关。
方法
分析了甲状腺缺损和用缺氧模拟剂(氯化钴)和抗坏血酸处理过的甲状腺癌细胞株(FTC-133和8305c)中HIF-1α和HIF-2α的表达以及维生素C的含量。
HIFs和缺氧诱导的葡萄糖转运蛋白的表达通过蛋白质印迹法确定,同时进行定量实时PCR(qRT-PCR)检测HIFs
mRNA水平。通过HPLC法测定抗坏血酸和脱氢抗坏血酸的水平。
结果
我们发现维生素C水平与HIF-1α呈负相关,但与甲状腺病变中的HIF-2α表达无关。这些结果与我们的体外研究一致,后者表明维生素C诱导甲状腺癌细胞FTC-133和8305C中HIF-1α的剂量依赖性降低,但不诱导HIF-2α蛋白质水平降低。
HIF-1α表达降低与甲状腺癌细胞中缺氧相关葡萄糖转运蛋白1(GLUT1)表达降低相关。
结论
结果表明,HIF-1α激活与甲状腺病变中的维生素C含量有关。我们的研究表明,高肿瘤组织抗坏血酸水平可能会限制HIF-1α及其靶标在甲状腺病变中的表达。
Expression of hypoxia inducible factor 1α and 2α and its association with
vitamin C level in thyroid lesions
Abstract
Background
Cells adapt to hypoxia by transcriptional induction of genes that participate in
regulation of angiogenesis, glucose metabolism and cell proliferation. The
primary factors mediating cell response to low oxygen tension are hypoxia
inducible factors (HIFs), oxygen-dependent transcription activators. The
stability and activity of the α subunits of HIFs are controlled by hydroxylation
reactions that require ascorbate as a cofactor. Therefore, deficiency of
intracellular vitamin C could contribute to HIFs overactivation. In this study,
we investigated whether vitamin C content of human thyroid lesions is associated
with HIF-1α and HIF-2α protein levels.
Methods
Expression of HIF-1α and HIF-2α as well as vitamin C content was analyzed in
thyroid lesions and cultured thyroid carcinoma cell lines (FTC-133 and 8305c)
treated with hypoxia-mimetic agent (cobalt chloride) and ascorbic acid. The
expression of HIFs and hypoxia–induced glucose transporters were determined by
Western blots while quantitative real-time PCR (qRT-PCR) was performed to detect
HIFs mRNA levels. Ascorbate and dehydroascorbate levels were measured by HPLC
method.
Results
We found an inverse correlation between vitamin C level and HIF-1α but not
HIF-2α expression in thyroid lesions. These results agree with our in vitro
study showing that vitamin C induced a dose - dependent decrease of HIF-1α but
not HIF-2α protein level in thyroid cancer cells FTC-133 and 8305C. The
decreased HIF-1α expression was correlated with reduced expression of
hypoxia-related glucose transporter 1 (GLUT1) in thyroid cancer cells.
Conclusion
The results demonstrate that HIF-1α activation is associated with vitamin C
content in thyroid lesions. Our study suggests that high tumor tissue ascorbate
level could limit the expression of HIF-1α and its targets in thyroid lesions.
Expression of hypoxia inducible factor 1α and 2α and its association with
vitamin C level in thyroid lesions | Journal of Biomedical Science | Full Text
https://jbiomedsci.biomedcentral.com/articles/10.1186/s12929-017-0388-y
Signaling hypoxia by hypoxia-inducible factor protein hydroxylases: a
historical overview and future perspectives
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5045067/
Potential Mechanisms of Action for Vitamin C in Cancer: Reviewing the
Evidence
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6037948/
The Hypoxic Tumor Microenvironment: A Driving Force for Breast Cancer
Progression - Europe PMC Article - Europe PMC
http://europepmc.org/articles/PMC4678039
缺氧性肿瘤微环境:乳腺癌进展的驱动力
格雷格·塞门扎*
抽象
肿瘤内缺氧是乳腺癌的常见发现,并且与转移风险和患者死亡率显着增加有关。缺氧诱导因子激活大量编码蛋白质的基因的转录,这些蛋白质促进原发性肿瘤血管形成和生长,基质细胞募集,细胞外基质重塑,转移前的利基形成,细胞运动,局部组织浸润,转移部位的外渗和维持产生继发性肿瘤所需的癌症干细胞表型。最近的临床前研究表明,细胞毒性化学疗法与抑制缺氧诱导因子的药物联合使用可改善三阴性乳腺癌女性的预后。
关键词:骨转移,肺转移,淋巴结转移,间充质干细胞,微囊泡,髓样抑制细胞,肿瘤相关巨噬细胞
去:
1.简介
1.1。肿瘤微环境对癌症进展有重大影响
三个一般过程对癌症的发生和发展有重大影响。首先,离散的体细胞突变导致肿瘤抑制功能丧失和癌蛋白功能丧失[1]。第二,表观遗传改变改变了基因表达的模式[2]。第三,肿瘤微环境的改变也导致基因表达的广泛变化[3]。已经高度关注了体细胞突变和表观遗传变化,这些突变很容易通过高通量分子技术进行研究[4]。相反,肿瘤微环境的变化较不易测定,但对癌症进展具有深远的影响。
肿瘤微环境可细分为:化学微环境,包括pH,PO2以及其他小分子(例如NO)和代谢产物(例如葡萄糖,谷氨酰胺,乳酸盐)的浓度;细胞微环境,包括肿瘤细胞,基质细胞以及这些细胞产生的细胞外基质(ECM)。基质细胞类型包括血管内皮细胞(EC)和周细胞,淋巴EC,成纤维细胞,肌成纤维细胞,以及各种骨髓来源的细胞,例如巨噬细胞,嗜中性粒细胞,肥大细胞,骨髓来源的抑制细胞(MDSC)和间充质干单元(MSC)。募集到原发性肿瘤的许多基质细胞促进原发性肿瘤的生长或转移[5]。
转移是在只有6%的女性患乳腺癌的初步介绍时发现,但女性在诊断早期疾病的30%最终将发展为转移性疾病,它负责每年4名万人死亡[6]。本次审查将集中在最近的进步在了解乳腺癌的演示化学微环境的一个重要方面怎么样,降低O2可用性(缺氧)的发病机制,发挥在与癌症进展收购产生深远的影响,即细胞微环境的主要影响侵入性和转移性导致患者死亡。事实上,肿瘤内低氧是驱动在基因表达的变化,从而导致在细胞改变的信令在许多癌症的病理性刺激比得上那些由体细胞突变或表观遗传变化。
1.2。乳腺癌和其他癌症中通常观察到肿瘤内缺氧
开创性的临床研究,其中在原位使用的Eppendorf微电极获得乳腺癌PO2测量,显示为65个毫米汞柱的位数(同所有的测量>
10个毫米汞柱)在正常乳腺组织;相反,对涉及200多个患者的10项研究进行的荟萃分析显示,治疗前乳腺癌的PO2中位数为10 mmHg
[7]。非常相似的结果在涉及超过700宫颈癌患者和500例头颈部癌症研究荟萃分析得到的;在这些癌症中,多项研究表明肿瘤内PO2
<10毫米汞柱之间的关联和降低的无病生存[7]。在这些研究中检测到的严重的肿瘤内低氧是扩散限制O2递送,其中快速癌细胞增殖导致过于远离血管细胞和灌注限制O2递送的结果,其中,在结构和功能异常肿瘤血管不保持恒定的血流量,从而使癌细胞甚至紧邻血管可能会缺氧[8]。坏死区域通常在晚期实体瘤中观察到,反映了在O2水平长时间维持不足以维持细胞生存力的后果[9]。
1.3。缺氧诱导因子的表达和活性在乳腺癌中增加
细胞通过缺氧诱导因子介导的基因表达变化来应对降低的O2利用率,缺氧诱导因子由O2调节的HIF-α亚基(HIF-1α,HIF-2α或HIF-3α)组成并组成性表达HIF-1β亚基[10,11]。
HIF-α亚基受到O2依赖的脯氨酰羟化,泛素化和蛋白酶体降解,在缺氧条件下受到抑制[12],从而导致蛋白质的稳定和快速积累以及转录活性。迄今为止,已经鉴定出超过1500种HIF靶基因,尽管在任何给定的细胞中,其中的数百种转录都会因缺氧而显着增加[13]。
肿瘤活检的免疫组织化学研究表明,HIF-1α蛋白水平升高与转移风险和淋巴结阳性[15],淋巴结阴性[16],HER2阳性[17],雌激素受体-增加有关。阳性[18]和未选[14,19]乳腺癌患者。
HIF靶基因编码的一组99 [20]或16 [21] mRNA在原发性乳腺癌中表达的增加也与患者死亡率的增加有关。
2.乳腺癌小鼠模型中HIF活性的后果
2.1。 HIF-1α促进原发性乳腺肿瘤生长和血管形成
在许多癌细胞类型中的研究已经确定,HIF-1α促进肿瘤异种移植物的生长和血管形成[22-24],这在由小鼠乳腺中表达多瘤中期T抗原(PyMT)驱动的乳腺癌的自体模型中也观察到肿瘤病毒(MMTV)启动子[25]。
HIF-1激活编码血管内皮生长因子的VEGF基因的转录[26],HIF-1α水平在乳腺癌中与VEGF表达和微血管密度相关,即使在原位导管癌(即乳腺癌的浸润前期)中发病机理[27]。
HIF介导癌细胞表达血管生成因子,例如VEGF,基质衍生因子1(也称为CXCL12)和干细胞因子(也称为试剂盒配体),它们诱导动员进入骨髓循环表达同源受体(分别为VEGFR2,CXCR4和CKIT)的血管生成细胞进入肿瘤并促进血管形成[28]。
2.2。 HIF-1α促进乳腺癌向腋窝淋巴结转移
在乳腺癌中,预测远处转移的最重要的临床发现是腋窝淋巴结受累的程度。相反,大多数具有远处转移的妇女都累及淋巴结,而且至少在某些情况下,乳腺癌有可能通过淋巴系统进入外周循环[29]。在将人类MDA-MB-231
[30]乳腺癌细胞(BCC)植入免疫缺陷小鼠的乳腺脂肪垫的原位小鼠模型中,表达短发夹RNA(shRNA)抑制HIF-1α,HIF-
2α,或两者同时导致原发肿瘤的淋巴管密度降低和转移至同侧腋窝淋巴结[31]。从在低氧条件下培养的BCC中收集的条件培养基可增加淋巴EC的迁移和增殖,而表达HIF-1α和HIF-2αshRNA会失去缺氧的作用[31]。
缺氧对体外淋巴内皮细胞迁移和增殖的影响需要血小板衍生生长因子B(PDGF-B)的HIF依赖表达,而PDGF-B敲除消除了。
PDGF-B的敲低还降低了体内淋巴管密度和淋巴结转移[31]。缺氧诱导HIF-1与PDGFB基因内含子3内的缺氧反应元件结合。值得注意的是,免疫组化显示,在1级乳腺癌活检中,HIF-1α和PDGF-B水平之间没有关联,但是在2级和3级癌症中观察到关联,这表明HIF-1α对于PDGFB是必需的但不充分乳腺癌进展过程中的基因表达[31]。
2.3。 HIF-1α促进乳腺癌细胞的骨定植
将MDA-MB-231细胞注射入免疫缺陷小鼠的左心室导致溶骨性骨转移的形成。与空载体亚克隆相比,HIF-1α的组成型活性或显性负型表达分别在长骨切片中增加或减少了肿瘤面积和血管密度[32]。
shRNA敲低HIF-1α还可以减少溶骨性病变的放射照相区域,减少骨转移中的血管密度,并增加注射BCC后的生存时间[33]。注射MDA-MB-231细胞后形成的骨转移灶含有低氧区域,脾细胞在体外暴露于低氧状态可抑制成骨细胞分化并刺激破骨细胞分化[32]。由于将BCC直接注射到循环系统中,因此这些研究没有提供令人信服的证据,表明HIF-1是从乳房到骨骼自发转移所必需的,而循环肿瘤细胞对骨骼定植的需求更为有限。
2.4。 HIF-1α是乳腺癌转移至肺部所必需的
已知将癌细胞离体暴露于缺氧状态会增加静脉注射后的肺部定植[34]。有条件地敲除乳腺上皮细胞中的HIF-1α可提高MMTV-PyMT小鼠的存活率,并使自发性肺转移数目减少约50%[25]。剔除人类MDA-MB-231
BCC中的HIF-1α,HIF-2α或两者,可显着降低乳腺脂肪垫注射后免疫缺陷小鼠的自发肺转移数量和总肺转移负担[35]。在此原位移植模型中,已描述了介导转移过程中特定步骤的大量HIF靶基因,如以下部分所述,为肿瘤内低氧对乳腺癌转移的影响提供了详细的分子基础。
3. Deconvoluting the multistep process of lung metastasis
3.1. Breast cancer cells recruit stromal cells to the primary tumor that promote
metastasis
The molecular mechanisms by which stromal cells are recruited to, and
communicate with, BCCs have not been well characterized. The production of
colony stimulating factor 1 (CSF-1; also known as macrophage colony stimulating
factor) by BCCs, has been reported to stimulate the recruitment of macrophages
to primary breast tumors, leading to the secretion by tumor-associated
macrophages (TAMs) of epidermal growth factor, which binds to its cognate
receptor on BCCs to stimulate invasion and metastasis [36–38]. However, the
mechanisms responsible for CSF-1 expression were not determined.
After subcutaneous co-injection of BCCs and MSCs, BCCs were reported to induce
MSCs to secrete the chemokine CCL5, which bound to its cognate receptor CCR5 on
BCCs to stimulate metastasis [39]. Remarkably, co-culture of human MDA-MB-231
BCCs with human bone marrow-derived MSCs for 48 hours prior to mammary fat pad
injection significantly increased metastasis to axillary lymph nodes and lungs
without having any effect on primary tumor growth [40].
Hypoxia induces the HIF-dependent expression of placental growth factor (PGF) by BCCs, which binds to VEGF receptor 1 on MSCs and promotes their recruitment to primary breast tumors [40]. MSCs express the chemokine CXCL10, which binds to its cognate receptor CXCR3 on BCCs, and BCCs produce the chemokine CXCL16, which binds to its cognate receptor CXCR6 on MSCs and promotes their recruitment to the primary tumor [41]. Expression of PGF, CXCR3, and CXCL16 by BCCs is induced by hypoxia in a HIF-dependent manner [40, 41]. In addition, CXCL10MSC → CXCR3BCC signaling stimulates CXCL16BCC → CXCR6MSC signaling and vice versa [41], providing a molecular basis for the stimulatory effect of co-culture. Thus, hypoxia induces a powerful feed-forward loop that amplifies signals for MSC homing (Fig. 1). In addition, CXCL10 was shown to enhance BCC migration and invasion [40]. The stimulatory effect of MSC co-culture on breast cancer metastasis was lost when HIF-1α/HIF-2α, CXCR3, or CXCL16 expression was knocked down by shRNA in BCCs [40, 41].
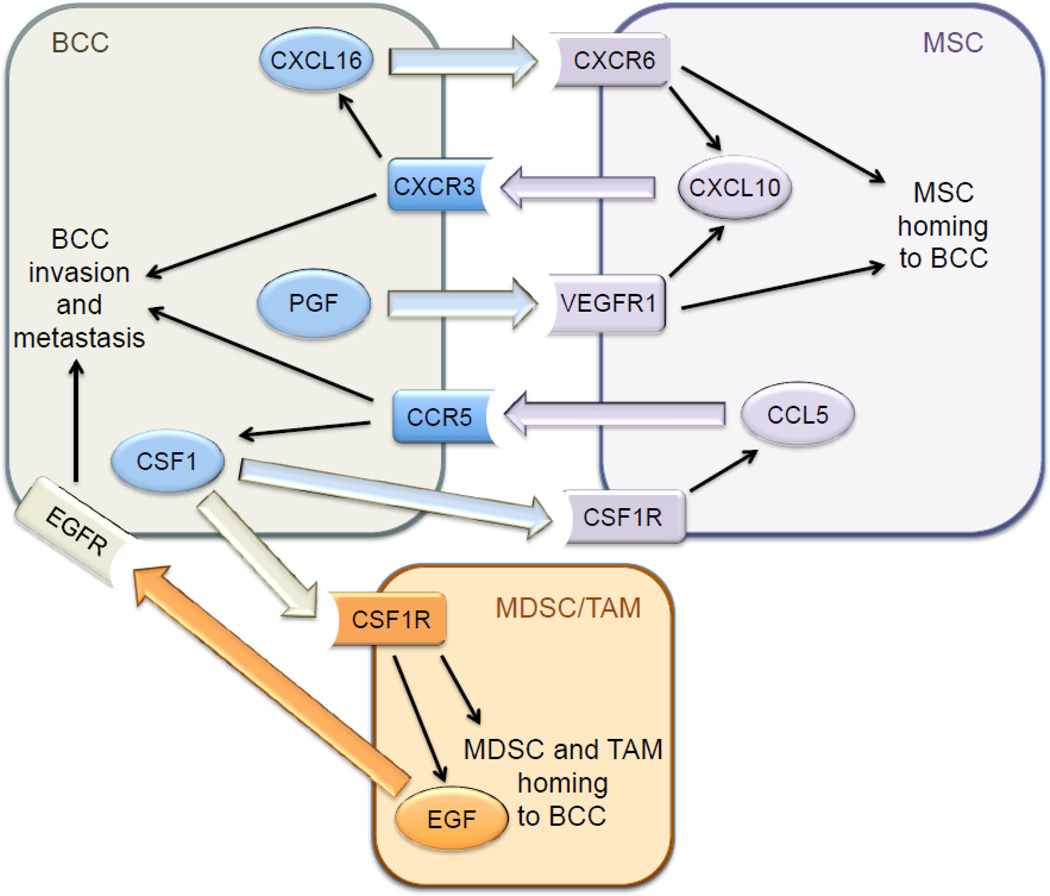
An external file that holds a picture, illustration, etc. Object name is nihms700440f1.jpg Open in a separate window Fig. 1 Signaling between breast cancer cells (BCCs), mesenchymal stem cells (MSCs), myeloid-derived suppressor cells (MDSCs), and tumor associated macrophages (TAMs) stimulates metastasis. Large colored arrows indicate intercellular signaling through interaction of ligand with its cognate receptor. Thin black arrows indicate intracellular signaling activated by ligand-receptor interaction. Hypoxia-inducible factors directly activate transcription of the genes encoding the three ligands (CXCL16, PGF, CSF1) and two receptors (CCR5, CXCR3) in hypoxic BCCs. The co-culture of BCCs with MSCs prior to mammary fat pad injection increased the recruitment of CSF1R+ F4/80+ TAMs and CD11b+ Ly6C+ MDSCs to the primary tumor in a HIF-dependent manner [41]. MSCs produce CCL5, which binds to its cognate receptor CCR5 on BCCs, and stimulates the production of CSF-1, which binds to its cognate receptor CSF-1R on MSCs, as well as on TAMs, thereby facilitating recruitment of TAMs to the tumor [38]. Hypoxia induces the HIF-dependent expression by BCCs of CCR5 and CSF-1, due to the direct binding of HIF-1 to two sites in the 5’-flanking region of the CSF1 gene and sites in the 5’-flanking region and exon 3 of the CCR5 gene [41]. CCL5MSC → CCR5BCC signaling stimulates CSF-1BCC → CSF-1RMSC signaling and vice versa, representing another feed-forward loop. Knockdown of HIF-1α/HIF-2α, CCR5, or CSF-1 expression in BCCs significantly impaired recruitment of TAMs and MDSCs to the primary tumor, and BCC metastasis to lymph nodes and lungs, without affecting primary tumor growth [41]. Thus, the recruitment of MSCs facilitates the subsequent recruitment of MDSCs and TAMs, with hypoxia stimulating homing of MSCs as well as MDSCs and TAMs to the primary tumor, leading to increased metastasis (Fig. 1). All of the experiments described above were performed using human MDA-MB-231 BCCs implanted into immunodeficient mice. To interrogate these pathways in the context of an intact immune system, 4T1 mouse mammary carcinoma cells were implanted into syngeneic (and immunocompetent) Balb/c mice. HIF-1α/HIF-2α double knockdown in 4T1 cells inhibited the recruitment of TAMs and MDSCs to primary tumors and the metastasis of BCCs to lymph nodes and lungs after mammary fat pad injection [41]. The in vitro and in vivo data described above indicated that CXCL10MSC → CXCR3BCC signaling induces CXCL16BCC → CXCR6MSC signaling and vice versa and that both CXCR3 and CXCL16 gene expression is induced by hypoxia in BCCs. Analysis of gene expression data from over 500 primary human breast cancers revealed that CXCR3 and CXCL16 mRNA levels were very highly correlated (P = 10-13, Pearson’s test) in the clinical specimens [41], suggesting that the hypoxia-induced signaling pathways delineated in BCC lines and mice are clinically relevant. 3.2. Hypoxia induces collagen hydroxylation that stiffens the extracellular matrix There is an extensive body of experimental data indicating that remodeling of the ECM promotes cancer invasion and metastasis [42, 43]. The histopathological detection of fibrosis, or increased fibrillar collagen content, in a primary breast tumor is associated with increased risk of cancer recurrence and patient mortality [44]. Type I collagen, which is the major constituent of ECM and the most abundant protein in the human body, forms when three procollagen polypeptides [two COL1(α1) subunits and one COL1(α2) subunit] assemble into a triple helix, as originally proposed by Pauling and Corey [45]. Type I collagen contains 338 X-Y-Gly triplets, with proline most commonly in the X position and hydroxyproline in the Y position preceding glycine [46]. The post-translational modification of proline residues in procollagen polypeptide chains to hydroxyproline is catalyzed by prolyl-4-hydroxylase (P4H) enzymes, which are α2β2 tetramers. In humans, there are three P4Hα isoforms and a single P4Hβ isoform, which are encoded by the P4HA1, P4HA2, P4HA3, and P4HB genes. Prolyl-4-hydroxylation is required for triple helix formation and extracellular deposition (Fig. 2). Expression of P4HA1 and P4HA2 was induced by hypoxia in several cell types [47].

Fig. 2
Collagen biogenesis. The formation of mature collagen fibers involves three
different types of post-translational modification that are mediated by prolyl
hydroxylase, lysyl hydroxylase, and lysyl oxidase family members. All of the
enzymes shown are products of genes that are transactivated by hypoxia-inducible
factors in hypoxic breast cancer cells.
X-Lys-Gly sequences in helical domains and X-Lys-Ala/Ser sequences in the
telopeptides of procollagens are also subject to lysine hydroxylation by
procollagen-lysine, 2-oxoglutarate 5-dioxgenase (PLOD) enzymes, which are
encoded by the PLOD1, PLOD2, and PLOD3 genes (Fig. 2). PLOD1 primarily
hydroxylates lysine residues in helical domains and PLOD2 modifies the
telopeptides, whereas PLOD3 hydroxylates type IV and V, but not type I, collagen
[48]. Like prolyl hydroxylation, lysyl hydroxylation occurs prior to collagen
deposition but, unlike prolyl hydroxylation, it is not required for
extracellular deposition of collagen; however, it is required for collagen
fibril assembly. Expression of PLOD1 and PLOD2 is induced by hypoxia [47].
After secretion, lysyl and hydroxylysyl residues are oxidatively deaminated by
lysine oxidase (LOX) and LOX-like proteins (LOXL1, LOXL2, LOXL3, LOXL4), which
is a modification that initiates the formation of intra-molecular as well as
inter-molecular crosslinks, which may involve other amino acids, including
histidinyl residues (Fig. 2). Collagen crosslinking is a major mechanism by
which ECM stiffness increases during cancer progression [49, 50]. Expression of
LOX, LOXL2, and LOXL4 was induced by hypoxia in several cell types [51–53].
Increased expression of P4HA1 and P4HA2 mRNA and protein was induced by exposure
of MDA-MB-231 cells to 1% O2 and knockdown of HIF-1α, HIF-2α, or both
significantly impaired the induction [54]. In breast tumors that arose after
MDA-MB-231 cells were implanted into the mammary fat pad of immunodeficient
mice, P4HA1 and P4HA2 co-localized with HIF-1α in perinecrotic (i.e. hypoxic)
regions. Knockdown of P4HA1 or P4HA2 expression significantly impaired primary
tumor growth and lymph node metastasis as well as completely eliminating lung
metastasis. The reduction in hematogenous metastasis was associated with
impaired tissue invasion and intravasation (invasion into blood vessels), as
measured by the number of circulating tumor cells [54]. In contrast, the number
of lung foci that developed after intravenous injection of P4HA1- or
P4HA2-knockdown cells was not significantly decreased, indicating that the
pro-metastatic effect of P4HA1 and P4HA2 reflects their roles in tissue invasion
and intravasation.
Collagen deposition and fiber formation, hydroxyproline content, and stiffness
were decreased in primary tumors derived from mammary fat pad implantation of
P4HA1- or P4HA2-knockdown cells. Although cancer-associated fibroblasts are
generally considered to be the major source of stromal collagen, these results
suggest that BCCs are also major contributors to ECM remodeling [54]. Treatment
of tumor-bearing mice with ethyl-3,4-dihydroxybenzoate, a P4H inhibitor,
modestly decreased primary tumor growth but dramatically reduced collagen
content in the primary tumor and metastatic burden in the lungs. Finally,
analysis of data from The Cancer Genome Atlas (TCGA) revealed that human breast
cancers with expression of P4HA1 or P4HA2 mRNA greater than the mean were
associated with significantly decreased patient survival [54], suggesting that
the preclinical studies described above have clinical relevance.
Expression of PLOD1 and PLOD2 mRNA and protein were induced by hypoxia in
MDA-MB-231 cells and induction was lost in cells with HIF-1α knockdown, whereas
HIF-2α knockdown had no significant effect [55]. Analysis of TCGA data revealed
that increased expression of PLOD2, but not PLOD1, in primary breast cancers was
associated with decreased patient survival. PLOD2 knockdown had no effect on the
growth or total collagen content of primary tumors, but decreased fibrillar
(crosslinked) collagen content and stiffness of primary tumors, and dramatically
reduced local tissue invasion as well as metastasis to lymph nodes and lungs
[55].
Analysis of LOX and LOXL1-4 expression in breast cancer cell lines revealed that
hypoxia induced LOX and LOXL4 expression in MDA-MB-231 cells, whereas LOXL2 was
induced in MDAMB-435 BCCs [56], and expression of LOX/LOXL proteins was not
detected in non-metastatic MCF-7 BCCs [53]. The hypoxic induction of LOX, LOXL2,
and LOXL4 was HIF-dependent. Analysis of primary breast cancers revealed a
heterogeneous pattern of expression with different tumors overexpressing LOX
only; LOX and LOXL2; LOXL2 and LOXL4; or LOX, LOXL2, and LOXL4 [53].
LOX family members are secreted proteins and conditioned medium from hypoxic
BCCs stimulated collagen crosslinking, which was lost when HIF-1α and HIF-2α
expression was knocked down [53]. Similar effects were observed when LOX or
LOXL4 was knocked down in MDA-MB-231 cells or LOXL2 was knocked down in
MDA-MB-435 cells [53]. Knockdown of LOX or LOXL4 in MDA-MB-231 cells or LOXL2 in
MDA-MB-435 cells inhibited lung metastasis [51, 53]. While the effect of LOX or
LOXL knockdown on metastasis is due in part to ECM remodeling of the primary
tumor, it is also due to remote ECM remodeling in the lungs, as described below.
3.3. The pre-metastatic niche is established by signals from the primary tumor
Sites of metastatic colonization in the lung are preconditioned for the arrival
of BCCs through modifications of the ECM and recruitment of bone marrow-derived
cells, a process known as pre-metastatic niche formation [57]. Formation of the
pre-metastatic niche is stimulated by factors that are secreted by the primary
tumor, including VEGF, PGF, tumor necrosis factor-α, transforming growth
factor-β (TGF-β), granulocyte colony stimulating factor, versican and LOX family
members, which stimulate pulmonary production of fibronectin and proinflammatory
proteins (S100A8 and S100A9) and recruitment macrophages, mast cells, and
neutrophils [58]. As described above, LOX and LOXL proteins deaminate the
ε-amino groups of lysine residues in collagen polypeptides, resulting in their
intramolecular and intermolecular crosslinking (Fig. 2). Since this process
occurs after extracellular deposition of collagens, LOX and LOXLs are secreted
proteins. In addition to modifying collagen in the primary tumor, LOX produced
in the primary tumor modifies collagen in the lungs, which facilitates the
recruitment of bone marrow-derived cells, which in turn facilitates the
recruitment of metastatic BCCs [59]. In mice implanted with MDA-MB-435 BCCs in
the mammary fat pad, increased collagen cross-linking was observed in the lungs
on day 8, whereas increased CD11b+ bone marrow-derived cells were first detected
in the lungs on day 16, and BCCs were first detected in the lungs on day 24,
demonstrating the sequential process of pre-metastatic niche formation [53].
Knockdown of LOX or LOXL4 in MDA-MB-231 or LOXL2 in MDA-MB-435 cells inhibited
pre-metastatic niche formation [53, 59]. Whereas anti-LOX antibodies or chemical
inhibitors of LOX (such as β-aminoproprionitrile) do not block the activity of
all LOXL proteins, the HIF inhibitors digoxin and acriflavine blocked
hypoxia-induced expression of all LOX and LOXL proteins, inhibited
pre-metastatic niche formation, and significantly reduced lung metastasis [60].
Taken together, these studies demonstrate that intratumoral hypoxia is an
important driving force for ECM remodeling that promotes BCC invasion and
metastasis through the HIFdependent activation of genes encoding collagen prolyl
hydroxylase, lysyl hydroxylase, and lysyl oxidase family members. MSCs were
reported to stimulate the production of LOX by BCCs [40, 61], providing another
example of crosstalk between these cell types that stimulates metastasis.
Finally, although we have focused on the role of LOX family members, it should
be noted that expression of other tumor-derived secreted factors that have been
implicated in pre-metastatic niche formation (e.g. PGF, VEGF, TGF-β) are also
induced by hypoxia in a HIF-dependent manner. 3.4. Activation of RhoA/ROCK1
signaling increases breast cancer cell motility Whereas cancer progression is
associated with remodeling that stiffens the ECM, the cytoskeleton undergoes
remodeling that transforms rigid, immobile, and polarized epithelial cells with
strong cell-to-cell adhesive contacts into motile and invasive cancer cells with
decreased cell-to-cell contacts and increased mesenchymal properties, a process
that is known as the epithelial-mesenchymal transition [62]. Members of the Rho
family of small GTPases play a major role in this process by mediating the
polymerization of actin (F-actin formation) to create stress fibers and the
phosphorylation of myosin to trigger contractility that is the molecular basis
for cell movement. Active (GTP-bound) Rho interacts with, and activates,
Rho-associated coiled-coil-forming kinase (ROCK), which mediates the
phosphorylation of myosin light chain (MLC), either directly or indirectly by
inactivating myosin phosphatase, leading to actin-myosin contraction [63, 64].
The force that is generated by actin-myosin contraction is used to pull on the
ECM at focal adhesions, leading to the activation of focal adhesion kinase
(FAK), with a positive auto-regulatory loop between RhoA and FAK signaling [65].
RHOA and ROCK1 gene expression is coordinately induced within the invasive
cancer cells of a breast tumor [66] and increased levels of RhoA and ROCK1
protein in biopsy specimens are associated with breast cancer progression [67,
68]. Somatic mutations are not the cause of RhoA or ROCK1 expression in most
breast cancers, leaving the underlying molecular mechanism undefined. Many
studies have demonstrated the effect of hypoxia on cell movement and invasion in
Boyden chambers [69, 70], which have many limitations including the confounding
effects of gravity and pore size and the inability to perform dynamic or single
cell analysis. Other studies have used video microscopy to study breast cancer
cells that were exposed to hypoxia, then replated and analyzed for only short
periods of time [71]. More recent studies have utilized microfluidic approaches
to image cell motility in three dimensional cell culture [72].
MDA-MB-231 subclones, which were stably transfected with empty vector (shEV)
or expression vectors encoding shRNAs targeting HIF-1α and HIF-2α (sh1/2α), were
plated on collagen-coated surfaces, exposed to 20% or 1% O2, and random motility
of single cells was dynamically monitored over 22 hours. Mean velocity and total
displacement of sh1/2α, but not shEV, cells increased significantly in response
to hypoxia [73]. Hypoxia induced stress fiber formation in shEV cells, which was
markedly impaired in sh1/2α cells. Molecular analyses revealed that RHOA and
ROCK1 mRNA expression was increased in metastatic (MDA-MB-231 and MDA-MB-435) as
compared to non-metastatic (MCF7 and T47D) BCC lines; hypoxia induced RHOA and
ROCK1 mRNA and protein expression in all BCC lines analyzed. Hypoxia also
induced phosphorylation of MLC and FAK in the metastatic cell lines, which was
blocked by treatment with the ROCK inhibitor Y-27632 [73]. Focal adhesion
formation was induced by hypoxia in shEV, but not sh1/2α, cells. The
hypoxia-induced increase in cell motility was lost in MDA-MB-231 cells with
knockdown of HIF-1α and HIF-2α or FAK [73]. Thus, hypoxia is sufficient to
activate RhoA → ROCK1 → MLC → FAK signaling, which is the basis for BCC
motility. Remarkably, the effect of hypoxia on motility was lost when cells were
plated on a soft substratum [73], thereby providing a direct connection to the
ECM remodeling (described in sections 3.2 and 3.3 above), which is also induced
by hypoxia (Fig. 3).
Fig. 3
Hypoxia induces collagen deposition and fiber formation that stiffen the
extracellular matrix (ECM). Hypoxia also induces RhoA/ROCK1-mediated myosin
light chain (MLC) phosphorylation and focal adhesion kinase (FAK) activity that
are required for actin-myosin contractility and cell motility on stiff ECM. Blue
oval, protein encoded by a HIF target gene; purple oval, protein that is
regulated by HIF-dependent RhoA/ROCK1 signaling.
Hepatocyte growth factor (HGF) stimulates cell migration by binding to its
cognate receptor MET, which is encoded by a HIF target gene [74]. Protein
tyrosine kinase 6 (PTK6) is a downstream effector of MET that is also encoded by
a HIF target gene and promotes breast cancer metastasis [75]. It would be
interesting to determine whether there is crosstalk between the HGF → MET → PTK6
and RhoA → ROCK1 → MLC → FAK signaling pathways in hypoxic BCCs.
3.5. Hypoxia promotes margination and extravasation of circulating tumor cells
Once BCCs have intravasated into a blood vessel, they are carried through the
bloodstream until their circulation is arrested by interaction with the luminal
surface of a vascular EC, a process that is known as margination. Exposure of
MDA-MB-231 cells to 1% O2 for 48 hours increased their adherence to vascular
ECs, due to the HIF-dependent activation of L1 cell adhesion molecule (L1CAM)
expression [35]. Knockdown of L1CAM markedly impaired adherence of BCCs to
vascular ECs ex vivo, reduced lung colonization after intravenous injection, and
decreased the number of spontaneous lung metastases after mammary fat pad
implantation [35].
BCC margination is followed by extravasation, which is the process of moving
from inside to outside of the blood vessel. In order for BCCs to migrate between
adjacent ECs, they secrete angiopoietin-like 4 (ANGPTL4), which binds to
receptors on ECs and inhibits EC-EC interactions [35, 76]. ANGPTL4 expression is
induced by hypoxia in a HIF-dependent manner [35]. Knockdown of ANGPTL4
expression in MDA-MB-231 cells had no effect on primary tumor growth but
dramatically impaired hypoxia-induced extravasation after intravenous injection
and lung metastasis after mammary fat pad implantation [35, 76].
3.6. Hypoxia induces the breast cancer stem cell phenotype
Once cells have invaded, intravasated, circulated, marginated, extravasated, and
homed to a metastatic niche, they must be capable of initiating a secondary
tumor. Only a small percentage of cancer cells within a primary tumor,
designated tumor-initiating cells or cancer stem cells (CSCs), have the capacity
to undergo asymmetric divisions in which a CSC divides, yielding one CSC and one
cell that has the ability to undergo a limited number of cell divisions that
will contribute to the exponential growth of the bulk tumor. CSC self-renewal
ensures that there will always be a source of cancer cells with proliferative
potential. Hypoxia was first shown to induce the CSC phenotype in glioma [77]
and, subsequently, in breast cancer [78, 79], in a HIF-dependent manner.
As for any cell type, the CSC phenotype is determined by patterns of gene
expression that are controlled by transcription factors. In the case of breast
CSCs, the co-activator TAZ (transcriptional activator with PDZ-binding motif)
has been shown to promote breast CSC self-renewal and tumor-initiation capacity
[80]. TAZ is an essential component of the Hippo pathway that regulates organ
mass in both invertebrates and vertebrates [81]. Phosphorylation by LATS1 or
LATS2 kinase maintains TAZ in the cytosol and prevents it from interacting with
DNA-binding proteins of the TEAD family to activate transcription. TAZ
expression is increased in 80% of high-grade breast cancers, which is associated
with amplification of the WWTR1 locus encoding TAZ in approximately 10% of
breast cancers [80], but the molecular basis for increased TAZ expression and
activity in the majority of TAZ-overexpressing breast cancers was not known.
Analysis of TAZ mRNA and protein expression revealed that that basal expression
was increased in the metastatic BCC lines MDA-MB-231 and MDA-MB-435 as compared
to non-metastatic BCCs, but TAZ expression was induced by hypoxia in a
HIF-1α-dependent and HIF-2α-independent manner in all BCC lines [82]. Chromatin
immunoprecipitation (ChIP) assays demonstrated hypoxia-induced binding of HIF-1α
and HIF-1β, but not HIF-2α, to a site in Intron 2 of the WWTR1 gene located 26
kb 3’ to the transcription start site. A 50-bp sequence containing the HIF-1
binding site functioned as a hypoxia response element in luciferase reporter
assays, demonstrating that WWTR1 is a direct HIF-1 target gene and implicating
intratumoral hypoxia as a mechanism for increased TAZ expression in high-grade
breast cancers. Hypoxia increased the expression of connective tissue growth
factor (CTGF) mRNA and increased the binding of TAZ to a region of the CTGF gene
promoter containing multiple TAZ/TEAD-binding sites (but no HIF-1 sites) in a
HIF-1α-dependent (and HIF-2α-independent) manner, indicating that HIF-1
amplifies TAZ target gene transcriptional activation in hypoxic BCCs by
increasing TAZ expression [82].
Analysis of TAZ by immunofluorescence or subcellular fractionation revealed
that hypoxia induced the nuclear localization of TAZ in MDA-MB-231 subclones
expressing a control shRNA or an shRNA targeting HIF-2α, but not in cells
expressing an shRNA targeting HIF-1α [82]. Immunoblot assays revealed
constitutive, low-level expression of LATS1, whereas LATS2 was abundantly
expressed in non-hypoxic BCCs [82]. Hypoxia induced the proteasomal degradation
of LATS2 through the HIF-1α-dependent expression of the ubiquitin protein ligase
SIAH1, which was shown to be a direct HIF-1 target gene (Fig. 4). Remarkably,
another study reported that the activity of YAP (Yes-associated protein), which
is a homolog of TAZ, was induced by HIF-1α-dependent expression of SIAH2, which
is a homolog of SIAH1, leading to LATS2 degradation [83].
An external file that holds a picture, illustration, etc.
.jpg)
Fig. 4
HIF-1 induces transcription of the WWTR1 and SIAH1 genes to increase TAZ
expression and activity in hypoxic breast cancer cells. WWTR1 encodes TAZ,
whereas SIAH1 encodes a ubiquitin protein ligase that targets LATS2 for
proteasomal degradation, thereby eliminating a negative regulator of TAZ nuclear
localization and transcriptional activity.
Breast CSCs are enriched among BCCs that express aldehyde dehydrogenase (ALDH+)
and among BCCs that form mammospheres when cultured on ultra-low adherence
plates [84, 85]. Hypoxia increased the number of ALDH+ and mammosphere-forming
CSCs, which was inhibited by shRNA knockdown of HIF-1α or SIAH1, but not HIF-2α,
whereas TAZ knockdown reduced breast CSCs under both hypoxic and non-hypoxic
conditions [82].
To analyze the effect of HIF or TAZ loss-of-function on tumor-initiating cells,
1000 cells of each MDA-MB-231 subclone were injected into the mammary fat pad of
immunodeficient female mice, which were scored for the presence of mammary
tumors 6 weeks later. Whereas 7 out of 7 mice injected with BCCs expressing the
non-targeting control shRNA developed tumors, tumors formed in only 3/7, 2/7,
and 0/7 mice injected with cells expressing shRNA targeting TAZ, HIF-2α, or
HIF-1α, respectively. These results are consistent with the role of TAZ and
HIF-1 in promoting the breast CSC phenotype, whereas HIF-2 may play either a
role in CSCs that is not reflected in the ALDH and mammosphere assays or a
non-CSC-related role that is required for early tumor formation, such as
promoting vascularization.
Analysis of gene expression and survival data from over 1,000 breast cancer
patients revealed that patients with expression of both HIF and TAZ target genes
in their primary tumor that was greater than the median had significantly
reduced survival compared to patients with low expression of HIF and/or TAZ
target genes [82]. These clinical results are consistent with a role for HIF-1
in amplifying TAZ target gene expression as well as the other (TAZ-independent)
molecular mechanisms described in previous sections by which HIF-1 promotes
breast cancer metastasis. Finally, in addition to the role of HIF-1 in mediating
increased TAZ activity, TAZ interacts with HIF-1α and serves as a co-activator
for HIF-1-dependent gene transcription in hypoxic BCCs [86], indicating that TAZ
and HIF amplify the activity of each other.
3.7. Microvesicles transfer the hypoxic phenotype to non-hypoxic cells and
stimulate their invasive and metastatic properties
Sections 3.1 to 3.6 have delineated molecular mechanisms by which hypoxia
induces the expression of genes that promote the invasive and metastatic
properties of BCCs. Extracellular vesicles are a novel cell-derived component of
the tumor microenvironment and recent studies suggest that hypoxic BCCs are able
to transfer their phenotype to non-hypoxic cells through the production of
microvesicles and exosomes, which are extracellular vesicles that contain
proteins, mRNAs, and microRNAs. Microvesicles are particles of 0.1- to 1-µm
diameter that form by outward budding and fission of the plasma membrane, which
is mediated by contraction of the actin cytoskeleton, whereas exosomes are 50-
to 200-nm diameter particles that form from inward budding of the limiting
membrane of multivesicular bodies and are released from the cell by fusion with
the plasma membrane [87–89]. In different cancers, extracellular vesicles have
been reported to promote angiogenesis, cancer stem cells, epithelial-mesenchymal
transition, or metastasis by effects on a variety of cell types in the tumor
microenvironment [90].
The small GTPases RAB27A and RAB27B have been implicated in exosome biogenesis
in melanoma and HeLa cells [91, 92], but the molecular mechanisms regulating
extracellular vesicle formation have not been delineated. Hypoxia stimulates the
release of extracellular vesicles that promote angiogenesis [93–97] and
increased exosome formation by hypoxic cancer cells requires HIF activity [96,
98], but the relevant HIF target genes have not been identified.
Analysis of peripheral blood from breast cancer patients revealed increased
numbers of microvesicles compared to controls [99]. Microvesicle formation was
increased in metastatic MDA-MB-231 and MDA-MB-435 cells as compared to
non-metastatic MCF-7 BCCs, whereas hypoxia induced microvesicle formation by
two-fold in all three BCC lines, but this effect was lost in subclones
expressing shRNAs targeting HIF-1α and HIF-2α [100]. Expression of RAB22A was
correlated with the expression of HIF target genes in primary breast cancers and
RAB22A expression greater than the median was associated with increased overall
survival and distant metastasis-free survival. ChIP assays revealed
hypoxia-induced binding of HIF-1 to a site in the 5’-untranslated sequence
within exon 1 of the RAB22A gene in BCCs [100].
Immunocytochemistry suggested that RAB22A was a component of the microvesicles
that formed in hypoxic cells and expression of shRNA targeting RAB22A eliminated
hypoxia-induced microvesicle formation as well as hypoxia-induced invasion of
BCCs through Matrigel [100]. Microvesicles collected from hypoxic BCCs and
incubated with non-hypoxic cells induced focal adhesion formation and invasion
in vitro, as well as lung colonization after intravenous injection, and this
effect was lost in cells expressing shRNA that targeted RAB22A. Knockdown of
RAB22A expression in MDA-MB-231 cells had no effect on primary tumor growth
after mammary fat pad injection but significantly reduced the number of
metastases from breast to lungs [100].
Previous studies implicated hypoxia-induced extracellular vesicle formation as a
means by which cancer cells influence the phenotype of vascular and other
stromal cells [93–96], whereas HIF- and RAB22A-dependent microvesicle formation
increases the invasive and metastatic properties of a primary breast tumor by
transferring the phenotype of hypoxic BCCs to non-hypoxic BCCs [100]. Many
questions remain to be answered about the role of extracellular vesicles in
cancer progression. Are tumor-derived factors, such as LOX family members, that
act to promote pre-metastatic niche formation delivered to the lungs in
extracellular vesicles? It would be interesting to test whether RAB22A
loss-of-function in BCCs affects collagen crosslinking in the lungs of
tumor-bearing mice. What are the molecular mechanisms by which hypoxia-induced
microvesicles affect focal adhesion formation, invasion, and metastasis? One
appealing mechanism is that the microvesicle cargo may include inducers of HIF-1
activity, such as microRNAs targeting negative regulators [97], or even HIF-1
itself [101].
3.8. Summary
HIFs function as master regulators of breast cancer metastasis by activating the
transcription of genes encoding proteins that participate in multiple steps of
the metastatic process, only a subset of which have been reviewed here (Fig. 5).
In addition to HIF target genes encoding proteins that directly participate in
metastasis, HIF-1 encodes transcriptional regulatory proteins such as JMJD2C,
which is required for breast cancer metastasis and encodes a histone demethylase
that is recruited by HIF-1 to target genes to further amplify their expression
[102].
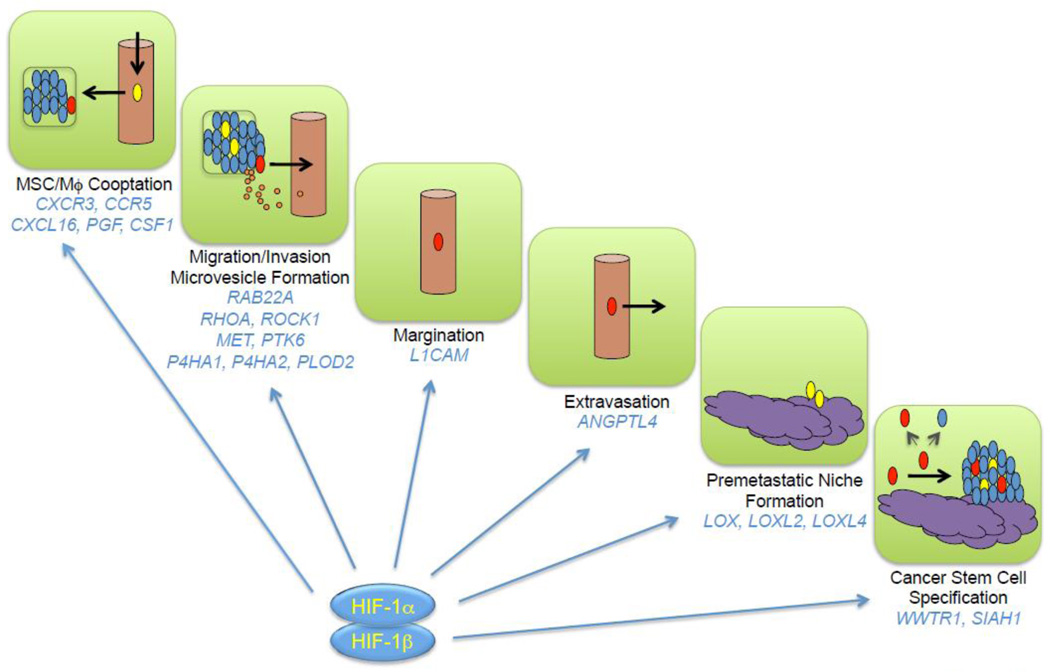
Fig. 5
HIF-1 activates the transcription of genes that control multiple steps in the
metastatic process. Blue, red, yellow, and orange ovals denote bulk cancer
cells, cancer stem cells, bone marrow-derived cells (mesenchymal stem cells,
myeloid-derived suppressor cells and tumor associated macrophages), and
extracellular vesicles (exosomes and microvesicles) respectively. Cancer stem
cells give rise to both stem cells and non-stem cells and can initiate secondary
(metastatic) tumors.
4. Implications for cancer therapy
Breast cancers are stratified into three major groups for therapy. Cancers that
express the estrogen receptor (ER) or progesterone receptor (PR) or both are
treated with an ER antagonist, such as tamoxifen, or an aromatase inhibitor,
such as letrozole. Cancers that overexpress human EGF receptor 2 (HER2) are
treated with an anti-HER2 antibody, such as trastuzumab, or a tyrosine kinase
inhibitor, such as lapatinib. Cancers that lack expression of ER, PR, or HER2,
which constitute approximately 15% of all breast cancers (30% among
African-American women), are designated “triple negative” and, in the absence of
any targeted therapy, are treated with cytotoxic chemotherapy, with greatly
increased rates of relapse, metastasis, and death compared to the other breast
cancer types [103]. Many of the studies described in this review were performed
with the MDA-MB-231 cell line, which was derived from a metastatic
triple-negative breast cancer [30].
There are now several drugs (acriflavine, digoxin, and ganetespib) that have
been shown to inhibit HIF-1 and shown to block breast cancer primary tumor
growth, angiogenesis, pre-metastatic niche formation, and local invasion as well
as metastasis to lymph nodes and lungs in orthotopic mouse models [31, 35, 60,
104]. Recent studies in mice indicate that combining digoxin with cytotoxic
chemotherapy (paclitaxel or gemcitabine) leads to tumor regression by blocking
HIF-dependent transcriptional responses that promote the resistance of breast
CSCs to chemotherapy [21]. HIF inhibitors may also induce immunogenic cell death
[105].
Analysis of hundreds of triple-negative breast cancers revealed a wide diversity
of somatic mutations none of which were found at high frequency [4]. Given the
remarkable overexpression of HIF target genes in triple-negative breast cancer
[4, 21], the master regulatory role of HIFs in metastasis (Fig. 5), and their
involvement in chemotherapy resistance, HIF inhibitors may represent a targeted
therapy for these breast cancers.
The Hypoxic Tumor Microenvironment: A Driving Force for Breast Cancer
Progression - Europe PMC Article - Europe PMC
http://europepmc.org/articles/PMC4678039
维生素C抗癌的作用机制:综述证据
Potential Mechanisms of Action for Vitamin C in Cancer: Reviewing the Evidence
Margreet C. M. Vissers* and Andrew B. Das
Author information Article notes Copyright and License information Disclaimer
Centre for Free Radical Research, Department of Pathology and Biomedical
Science, University of Otago, Christchurch, Christchurch, New Zealand
Edited by: Jin Wang, Fudan University, China
Reviewed by: Simona Martinotti, Università degli Studi del Piemonte Orientale,
Italy; Venkat R. Pannala, Biotechnology HPC Software Applications Institute
(BHSAI), United States
*Correspondence: Margreet C. M. Vissers, zn.ca.ogato@sressiv.teergram
This article was submitted to Oxidant Physiology, a section of the journal
Frontiers in Physiology
抽象
数十年来,人们一直在争论维生素C(抗坏血酸)是否具有抗癌作用。癌症患者已在不受监管的环境中以饮食补充剂或通过输注给药的药理剂量使用抗坏血酸,有许多临床益处的报道,但缺乏严格的临床试验数据。由于缺乏对影响有效剂量,给药时间和可能的反应性癌症模型选择的作用机理的了解,阻碍了适当临床试验的设计。最近,对抗坏血酸生物活性的广泛了解导致了许多有关抗癌活性机制的假说。其中最突出的是通过超生理浓度的抗坏血酸的自氧化产生大量的过氧化氢,并刺激了具有抗坏血酸辅助因子的2-氧戊二酸依赖性双加氧酶家族的酶(2-OGDDs)。假定过氧化氢的产生会产生优先针对癌细胞的氧化应激。
2-OGDD包括调节缺氧反应的羟化酶,肿瘤存活,血管生成,干细胞表型和转移的主要驱动力,以及表观遗传的组蛋白和DNA脱甲基酶。后者特别受关注,最近的研究表明抗坏血酸在血液癌症中调控十一个十一个转位酶(TET)DNA脱甲基酶的作用。对这些拟议机制的支持来自许多体外研究,异种移植动物模型始终显示出抗坏血酸给药的抗癌作用。但是,尚未从体内获得任何特定作用机制的决定性证据。目前正在进行许多早期临床试验,需要潜在的作用机理证据来为最合适的研究设计和癌症模型选择提供依据。希望这些信息将产生可靠的临床数据,从而避免为这个已经引起争议的辩论增加任何进一步的争议。
关键词:抗坏血酸,低氧诱导因子,过氧化氢,铁介导的自氧化,抗氧化剂,表观遗传脱甲基酶,十一个转位酶,HIF羟化酶
介绍
几十年来,维生素C是否在癌症的发展和调节中发挥作用一直是研究和讨论的话题。有大量文献记录了抗坏血酸在体外和体内的潜在抗肿瘤作用,其中许多报道了对癌细胞的细胞毒性和动物模型中肿瘤生长的减慢(有关综述,请参见Du等人, 2012; Park,2013; Fritz等人,2014; Wilson等人,2014; Mata等人,2016; Cimmino等人,2018; Klimant等人,2018)。然而,人类临床研究很少,最近的I / II期研究旨在确定抗癌药物药理学剂量对晚期癌症患者的耐受性(Hoffer等,2008,2015; Stephenson等,2013)。 。其中一些研究表明,高剂量抗坏血酸治疗可能对胰腺癌(Monti等人,2012; Cieslak和Cullen,2015)和其他晚期癌症(Hoffer等人,2015)的患者具有临床益处,但治疗的可能性太低迄今为止,已在这些研究中对患者进行了分析。此外,饮食中维生素C的使用已与改善乳腺癌患者的预后相关(Harris等,2013,2014)。
对抗坏血酸用于癌症的兴趣始于1970年代,当时Pauling和Cameron观察到每天用约10 g抗坏血酸静脉滴注治疗的晚期癌症患者的生存期明显延长,因此提倡使用抗坏血酸(Cameron and Pauling,1976, 1978)。当时,许多人都对这些主张表示怀疑,这主要是因为有关抗坏血酸生物活性的现有信息无法与合理的抗癌机制相吻合。有人提出抗坏血酸缺乏症可能会影响结缔组织,尤其是胶原蛋白的合成(McCormick,1959; Cameron和Pauling,1973),但没有解释可以解释静脉内给药的必要性,因为缺乏症可以很容易地克服。饮食补充。
梅奥诊所试图在患有晚期癌症的患者中重复Pauling和Cameron在一项随机安慰剂对照研究中的结果,但是他们在每天口服10 g维生素C后未能发现任何临床益处(Moertel等,1985)。现在认为这两个数据集之间的根本区别是抗坏血酸盐的口服与静脉内途径,已知这会导致维生素的药代动力学显着改变(Padayatty和Levine,2001; Padayatty等,2004)。当与对肿瘤生物学的更深入的了解一起观察时,有关维生素C的药代动力学和功能的信息导致了有关抗癌活性潜在机制的新假设的发展。这些关键信息在1970年代尚不可用,但现在重新考虑了抗坏血酸在癌症中的潜在作用。
高剂量抗坏血酸输注是医师和辅助及替代药物提供者都易于使用的疗法,但没有强有力的临床疗效证据(Padayatty等,2010)。公认需要进行良好的临床研究以确定这种做法的抗癌功效并建立适当的临床指南。病例报告表明,在某些情况下维生素C可以提供临床益处(通常是静脉输注大剂量药物,几个月内服用一个疗程)(Drisko等人,2003;
Padayatty等人,2006; Mikirova等人) (2016年;
Raymond等人,2016年)和最近的I期试验表明,大剂量维生素C是化疗的有用辅助剂(Welsh等人,2013年;
Hoffer等人,2015年)。但是,鉴于癌症患者普遍使用抗坏血酸,因此临床优势并不普遍,这表明只有一部分患者可以受益。这凸显了一个问题:如何确定潜在的反应患者并确定最佳治疗方案,剂量和干预频率?为了解决这些问题,我们必须了解抗坏血酸在癌症中的作用,这一点至关重要。当前,正在研究许多假设,在本综述中,我们将考虑迄今为止支持这些假设的证据,并根据我们对抗坏血酸化学和生物学的了解进行评估。
抗坏血酸的吸收和代谢
抗坏血酸的吸收和代谢抗坏血酸是一种小的,高度水溶性的分子,来源于葡萄糖,存在于所有动植物中(Hornig,1981; Smirnoff and
Wheeler,2000; Padayatty and
Levine,2016)。大多数动物在肝脏或肾脏中合成它,但它已成为人类,其他灵长类动物,豚鼠和果蝠的维生素,它们在古龙内酯氧化酶(生物合成途径中的末端酶)中发生了突变(Chatterjee等,1975;
Banhegyi等人,1997; Linster和Van Schaftingen,2007;
Du等,2012)。在所有动物中,抗坏血酸通过循环运输到组织,大多数细胞通过钠依赖性维生素C转运蛋白SVCT1和SVCT2的主动摄取,将维生素浓缩至血浆水平(平均约50μM)的许多倍。
1997; May和Qu,2005; Savini等,2008; Harrison和May,2009; Mandl等,2009;
Nualart等,2014)。例外是红细胞,它不表达SVCT2并通过GLUT吸收脱氢抗坏血酸(DHA)来积累抗坏血酸(Tu等人,2017)。组织水平差异很大,某些器官,尤其是脑,肾上腺,肝脏和白细胞,其浓度高达20
mM(Tsao,1997)。人们认为高的细胞内水平反映了对抗坏血酸作为必需的酶辅因子的需求(Tsao,1997; May和Qu,2005)。
高水溶性和抗坏血酸在体内的主动运输意味着该化合物易于获取和分配,但不能储存,并且营业额恒定。因此,如果不能保持足够的摄入量,那么对人类饮食摄入的依赖会导致缺乏,这与抗坏血酸补充剂在癌症中的作用有关。在患病期间,营业额似乎会加速(Bonham等人,1999; Fain等人,2003; Gan等人,2008; Evans-Olders等人,2010),尽管信息稀少,但许多癌症患者当进行测量时,发现抗坏血酸缺乏(Anthony and Schorah,1982; Ray and Husain,2001; Mayland et al。,2005; Shah et al。,2009; Badid et al。,2010; Nagamma et al。, 2014)。这些低血浆水平可能与较低的组织水平相关。这已在白细胞中证实(Levine等,2001),在抗坏血酸依赖性小鼠模型中,低血浆水平导致几乎完全组织缺乏(Vissers等,2011)。相比之下,已知合成抗坏血酸的动物可在不受健康挑战的情况下维持血浆饱和水平,从而显着提高产量以补偿生病时的周转率增加(Chatterjee等,1975; Michels和Frei,2013; Campbell等,2015)。 。这是使用动物肿瘤异种移植模型进行实验时要考虑的重要点。除非研究是用豚鼠或诸如Gulo-/-基因敲除小鼠这样的突变动物进行的,否则通过内源性合成可以将血浆和组织抗坏血酸水平维持在较高的基线水平,补充可能影响很小。野生型动物和饲养在野生型背景上的基因敲除动物(例如Gulo-/-小鼠)也表现出通过肠道吸收抗坏血酸的能力有限(Michels和Frei,2013)。因此,如果将这些动物用于实验环境,则膳食补充剂的作用可能有限,并且应始终监测血浆和组织水平,以确认抗坏血酸状况和该模型对人类疾病的适用性。应当指出的是,这种测量很少进行,这会使公开的实验数据的解释变得复杂。
抗坏血酸的化学和功能
抗坏血酸的化学性质决定了它的生物活性。可能以其作为抗氧化剂的能力而广为人知:此属性反映了其易于进行单电子或双电子氧化的能力,分别产生相对稳定的抗坏血酸基或DHA。
DHA在中性pH下不稳定,除非在体内被谷胱甘肽或硫氧还蛋白还原以再生还原的抗坏血酸,否则DHA会迅速分解为二酮古洛糖酸,草酸和苏糖酸(Washburn和Wells,1999;
Smirnoff,2000)。人体通过肾脏(Washko et al。,1992,1993; Washburn and Wells,1999; Linster
and Van
Schaftingen,2007)。因此,DHA仅占体内抗坏血酸的很小一部分,并且不可能以高于1-2μM的浓度存在(Dhariwal等,1991;
Michels和Frei,2013; Pullar等,2018)。
。在分析血液或组织时,体内的存在量通常可能被高估了,因为似乎大多数检测到的DHA可能在样品处理过程中反映了空气中的氧化(Dhariwal等,1991;
Levine等,1998; Dhariwal等,1991)。 Michels和Frei,2013;
Pullar等,2018)。抗坏血酸的氧化和DHA的分解很可能是造成每日营业额的原因,并且在患病期间可能会加剧(Chatterjee等,1975;
Bonham等,1999; Fain等,2003; Gan等) ,2008; Evans-Olders等,2010)。
抗坏血酸能够螯合和还原过渡金属离子,特别是Fe3 +和Cu2 +(Du等人,2012;
Padayatty和Levine,2016),并且该特性有助于其促进饮食中铁的吸收(Lane和Richardson,2014)。 )。它还可以通过Fe2
+的循环(方程式1)实现促氧化剂活性,并且在氧气存在下会通过促进Fenton反应而导致生成超氧化物,H2O2和高反应性氧化剂,例如羟基自由基(方程式1)。
2)和Haber-Weiss化学(Winterbourn,1981; Buettner,1987; Smirnoff,2000; Koppenol,2001;
Levine等人,2011; Michels and Frei,2013)(图图11)。
图1
抗坏血酸的抗氧化剂活性。抗坏血酸与氧或游离过渡金属离子(如Fe2 + / 3 +或Cu + / 2
+)的反应可导致生成H2O2,而H2O2本身具有细胞毒性,可进一步反应以加剧氧化应激(Shah等人,2009年; Vissers等,2011;
Nualart等,2014)。
AscH– + Fe3 +→Asc•– + Fe2 ++ H +(1)
Fe2 ++ H2O2 + H +→Fe3 ++ H2O + HO•(2)
还原过渡金属的能力可能是抗坏血酸盐活性的基础,而抗坏血酸盐是许多含铁和含铜酶的必需辅助因子。在这种能力下,它可以确保通过还原状态下的活性位点金属来优化酶的活性(Kivirikko等,1989;
Rebouche,1991; Ozer和Bruick,2007; Mandl等,2009; Grosso等等人,2013; Vissers等人,2014)。
抗坏血酸的抗癌活性
正在研究抗坏血酸盐的所有已知特性,以研究其潜在的抗癌活性。有大量研究表明,抗坏血酸酯单独或与化学治疗药物联合使用对体外肿瘤细胞具有细胞毒性作用(Wells等,1995;
Reddy等,2001; Pathak等,2002; Wozniak和Anuszewska) ,2002; Guerriero等,2006;
Kassouf等,2006; Chen等,2007,2012; Martinotti等,2011; Verrax等,2011; Ma等,2014;
Cieslak等, (2015年; Xia等人,2017年)或辐射(Herst等人,2012年;
Castro等人,2014年)。许多这些研究的细胞毒性反映了当抗坏血酸浓度为1
mM或更高时,细胞培养基中产生的H2O2产生的氧化应激,表现为细胞周期停滞增加,p53上调,ATP含量降低,线粒体功能受损,通过凋亡抑制抗氧化剂基因表达NrF-2和/或细胞死亡(Tarumoto等,2004;
Fromberg等,2011; Rouleau等,2016; Yang等,2017)。抗坏血酸水平远低于1
mM也已显示出抗癌作用:低至100μM甚至1μM的抗坏血酸水平可增强癌细胞对依托泊苷,顺铂或阿霉素的敏感性(Kurbacher等,1996) ;
Reddy等,2001; Tarumoto等,2004;
An等,2011)。在这些低浓度下的作用机理仍不清楚,但可能涉及涉及p53的细胞存活途径的修饰(Kurbacher等,1996; Reddy等,2001;
Tarumoto等,2004; An等,2011)。 。
在这些体外研究中,高度可变的实验设计和结果差异突显了研究的复杂性,涉及向细胞培养系统中添加抗坏血酸,从而可能导致伪影和实际细胞功能的改变。抗坏血酸与化学治疗药物的相互作用可能反映了细胞外抗坏血酸的氧化还原特性和许多细胞内辅助因子的活性。临床上也担心抗坏血酸可能会阻碍化疗药物的作用(Ludke等,2009,2010;
Perrone等,2009),但是这一领域仍然知之甚少。有一个重要的共识,即抗坏血酸可以与吉西他滨协同作用(Kassouf等人,2006;
Espeey等人,2011; Martinotti等人,2011; Volta等人,2013;
Cieslak等人,2015)和治疗方法两种药物联合使用在最近的I / II期试验中未引起任何不良事件(Monti等,2012; Welsh等,2013)。
考虑抗坏血酸和化学治疗药物之间的所有潜在相互作用不在本综述的讨论范围内,但应注意的是,在考虑将抗坏血酸引入临床癌症时,对这一领域的明确了解将非常重要。相反,我们旨在考虑最新假设的证据,这些假设表明抗坏血酸的抗癌活性反映了其氧化还原,促氧化剂或酶辅因子的活性(图22)。大量的文学作品正在检验这些想法,我们将考虑当前证据在多大程度上支持这些机制。
图2
现有假设的概述,可能会通过抗坏血酸促进抗癌活性。
小鼠异种移植模型
小鼠肿瘤异种移植模型的使用在许多研究中很普遍,并且在报告的结果中具有高度的一致性:通过饮食或通过大剂量腹膜给药来补充补充抗坏血酸盐会导致生长速率降低,并且(Gao等,2007;
Chen等,2008; Cha等,2011,2013; Espey等,2011; Ma等,2014; Campbell等,2015,2016b) ; Yun
et
al。,2015)。尽管肿瘤模型和抗坏血酸的剂量方案有所不同,但数据的相似性增强了对具有抗癌功能的抗坏血酸的支持,但在大多数情况下,其作用机理是从体外获得的数据推断出来的。很少有研究报道肿瘤生物学方面的变化,这些变化允许深入了解作用机理和结论,而不仅仅是肿瘤生长与抗坏血酸利用率之间的简单关联。没有这些信息,来自动物研究的体内数据将无法直接检验有效的假设,下面将对此进行考虑。
抗坏血酸作为前氧化剂
静脉输注抗坏血酸会导致血浆浓度在mM范围内,尽管是短暂的(半衰期约2小时)(Padayatty等,2004),这一认识导致人们提出抗坏血酸的自氧化和/或对金属的刺激。催化性活性氧的产生可以产生大量的H2O2,这些H2O2对癌细胞具有选择性毒性(Carosio等,2007;
Belin等,2009)。在组织培养基中,已知抗坏血酸盐浓度超过1 mM具有细胞毒性(Buettner,1988; Miller等,1990;
Halliwell等,2000;
Chen等,2005,2007,2008)。当将铁螯合剂添加到培养基中时,抗坏血酸自氧化的速度变慢,过氧化氢酶可以抑制细胞毒性,支持了这种现象对H2O2生成的依赖性(Halliwell等,2000;
Clement等,2001; Wee等人,2003;
Chen等人,2007)。癌细胞已被证明比原代细胞系更易受H2O2和氧化应激的影响,这与H2O2的代谢和抗氧化能力有关(Doskey等,2010;
Du等,2010; Erudaitius等,2010 ; Olney等,2013;
Schoenfeld等,2017)。与组织培养基相反,血浆和中性pH下抗坏血酸的自氧化反应缓慢(Buettner,1988),但是在生物流体中检测到了抗坏血酸基团,其中可能含有痕量的催化铁(Buettner
and Chamulitrat,1990; Chen等)。等(2007)。
如上所述,体外抗坏血酸介导的对细胞的细胞毒性已经超过了诱导大量H2O2的1 mM阈值(Chen等,2011; Espey等,2011; Fromberg等, 2011; Rouleau等,2016; Kim等,2018)。这些观察结果推动了包括静脉给药抗坏血酸药理学剂量的临床试验研究和一期临床试验,这些药物的目的是在肿瘤的循环和细胞外液中达到mM水平(Hoffer等,2008,2015; Monti等,2012; Stephenson等,2013; Welsh等,2013)。尽管有建议从这些维生素C输注中获得临床益处(Hoffer等人,2008,2015; Stephenson等人,2013; Welsh等人,2013),但确定这是否是由于维生素C的摄入仍然是一个挑战。抗坏血酸介导的抗氧化剂作用。尚不清楚体内的肿瘤中是否以足够的浓度生成过氧化氢以破坏癌细胞。鉴于抗坏血酸的自氧化高度依赖于氧气的可用性,围绕缺氧肿瘤环境对H2O2产生的影响存在一个问题。据我们所知,由体内超生理抗坏血酸浓度引发的H2O2介导的细胞毒性的确凿证据,例如在原位暴露于高剂量抗坏血酸的肿瘤中氧化损伤的检测,尚无定论。
癌细胞摄取DHA和相关的氧化应激
脱氢抗坏血酸在结构上与葡萄糖相似,可以通过GLUT吸收到细胞中,这可能有助于红细胞,感染部位的嗜中性粒细胞和身体其他部位的细胞内池(Rumsey等,1997;
2000; Corpe等人,2005; Wilson,2005;
Michels和Frei,2013)。一旦进入细胞,DHA就会被GSH,NADH和NADPH依赖性酶还原,从而潜在地耗尽细胞中的这些关键分子(May等人,1997,1999;
May,2002,2011)。最近有人提出了这种现象与KRAS和BRAF突变细胞中GLUT1上调之间的联系,以解释抗坏血酸在结直肠癌中的抗肿瘤活性(Yun等人,2015)。
KRAS和BRAF突变在大肠癌中很常见,并且与GLUT1的上调和糖酵解表型有关。当与mM浓度的抗坏血酸盐孵育时,具有KRAS和BRAF突变的培养癌细胞通过GLUT1吸收DHA,这与细胞活力的丧失有关。将结果外推到植入了突变型KRAS和BRAF癌细胞的体内小鼠模型中,当通过每天腹膜内注射4
g /
kg给予动物高剂量的维生素C时,肿瘤的生长会减慢。这项全面研究中的数据清楚地表明,GLUT1的表达上调可导致这些癌细胞系快速吸收DHA,但关于它与所观察到的细胞毒性之间的因果关系的不确定性要小得多。如上所述,在所用抗坏血酸浓度为1-2
mM时,通过自氧化在培养基中生成H2O2可能同样导致GSH耗竭和细胞毒性。可以通过在培养基中加入过氧化氢酶(例如过氧化氢酶)来消除这种可能性,并且在没有此信息的情况下,仍然不确定细胞死亡是否是由于摄取DHA和随后的细胞内氧化还原应激所导致的(如所提议的),或者是否是由于介质中生成的H2O2。未测试维生素C处理的小鼠中KRAS突变的肿瘤生长抑制是否与肿瘤中的氧化应激标志物相关。据报道,用他莫昔芬治疗的KrasG12D小鼠的抗坏血酸水平较高,而肠道息肉较少,但未见此突变的小鼠,但尚不确定这些息肉是否显示较高的氧化应激水平。在培养的KRAS和BRAF突变细胞中,糖酵解的代谢标记物降低了,尽管这可能与所研究的假设相符,但其同样可能受到低氧表型下调或H2O2介导的毒性的影响。
如前所述,DHA在中性pH下不稳定,通常在血浆或组织样品中未检测到(Dhariwal等,1991; Michels和Frei,2013;
Pullar等,2018)。因此,当考虑通过GLUT吸收DHA有助于抗坏血酸的抗肿瘤活性时,重要的是要证明DHA在体内以足够高的浓度存在,能够与葡萄糖的生理浓度竞争。有趣的是,最近报道了抗坏血酸可增强西妥昔单抗在KRAS突变型结肠癌细胞系中的细胞毒性,并同时逆转糖酵解的Warburg表型(Aguilera等,2016)。同样,在另一项研究中,这种活性与SVCT2依赖性抗坏血酸的细胞蓄积有关(Jung等,2016)。总之,这些研究描绘了抗坏血酸与结肠癌细胞相互作用的复杂情况。
低氧表型的下调
在发展中的肿瘤中,快速的细胞分裂和不良的血管形成会导致局部区域的氧气和营养物质被剥夺,从而导致HIF的激活。这些转录因子决定了肿瘤是否成功适应了压力微环境,从而驱动了涉及糖酵解和葡萄糖转运,血管生成,转移以及对化学疗法和放射疗法的抵抗力的众多基因的转录(Semenza,2010,2016;
Ratcliffe,2013)。高HIF活性已显示可促进乳腺癌中干细胞表型的表达(De Francesco等人,2015;
Semenza,2015,2016),并且与许多癌症的预后不良有关(Dachs和Tozer, 2000; Vleugel等,2005; Cao等,2009;
Semenza,2010,2015,2016; Volinia等,2012; Wang等,2012,2014; Zhang等,2012; Deb等(2014);
Liu等人,2014; Li等人,2016; Schoning等人,2017)。因此,HIF现在被认为是癌症治疗的重要靶标。
HIF的激活受脯氨酸和天冬酰胺酰羟化酶(HIF羟化酶)控制,这些酶修饰调节性HIF-α亚基(Bishop和Ratcliffe,2014年;
Pugh和Ratcliffe,2017年),将蛋白质靶向于蛋白酶体降解并阻止蛋白质的形成。活性转录复合物。
HIF羟化酶属于含铁的双加氧酶家族,利用氧气和2-氧代戊二酸酯作为底物(Ozer和Bruick,2007年),抗坏血酸作为辅因子起作用,可能是通过在还原态州(Koivunen等,2004)。辅助因子的作用是抗坏血酸的特异作用(Myllyla等,1978),其结构上对羟化酶活性位点具有特异性(Kaczmarek等,2009)。当细胞缺乏抗坏血酸时,HIF羟化酶活性受到损害,HIF转录活性增加,特别是对轻度或中度缺氧条件的响应(Knowles等人,2003;
Vissers等人,2007; Kuiper等人,2014a ;
Campbell等人,2016a)。这些观察结果提出了这样的假设:增加对癌细胞的抗坏血酸供应可以刺激HIF羟化酶的活性并降低HIF的活化,从而减慢肿瘤的生长速度。
与此假设相符的证据越来越多。对维生素C依赖性Gulo-/-小鼠的研究表明,在饮食中摄入可变剂量或腹膜内注射抗坏血酸药物后,体内抗坏血酸含量,HIF活化与体内肿瘤生长之间存在很强的联系(Campbell等人,2015,2016a
)。对荷瘤的Gulo-/-小鼠每日高剂量维生素C的给药表明,HIF-1转录活性与抗坏血酸摄取有关而被抑制。腹膜内输注后,肿瘤抗坏血酸含量的升高是短暂的,并且每天(而非隔日)给药维持水平。
HIF-1表达降低和肿瘤生长减慢也需要每天给药,抗坏血酸水平与肿瘤微血管密度降低和缺氧区域减少有关,表明整个肿瘤的氧输送更加有效(Campbell等,2016b)。
对人类肿瘤组织的回顾性分析发现,肿瘤中抗坏血酸的含量与子宫内膜癌,结直肠癌和甲状腺癌组织中HIF激活的标记呈负相关(Kuiper等,2010,2014b;
Jozwiak等,2015)。
。较高的肿瘤抗坏血酸与大肠癌患者的无病生存期增加相关(Kuiper等人,2014b)。综上所述,这些数据为抗坏血酸介导的HIF转录活性下调提供了有力的证据,这可能与较慢的肿瘤生长相一致。
表观遗传机制
遗传和表观遗传学的改变与癌症的发展密不可分(Baylin和Jones,2011,2016)。癌症中最引人注目的表观遗传变化之一是整体DNA甲基化不足,它可能导致基因组不稳定和染色体易碎性增加(Berman等,2011;
Ehrlich and
Lacey,2013)。此外,低甲基化可以激活转座因子和致癌基因的转录,从而提供多种机制来促进肿瘤发生。除了总体的低甲基化,还建立了定位于肿瘤抑制基因启动子的高甲基化(Baylin和Jones,2011)。相反,大多数含有CpG岛的基因启动子在成体干细胞中并未甲基化,这对于这些基因保持活性或准备被激活至关重要(Shen和Laird,2013)。这些发现对于表皮遗传学改变是可逆的,而对于癌症驱动突变本身更难以靶向和逆转,因此对于癌症疗法的发展是令人鼓舞的。
从细胞的正常功能到癌症的治疗,抗坏血酸在表观遗传学中的作用受到了越来越多的关注(Lorsbach等,2003; Monfort和Wutz,2013;
Young等,2015; Camarena和Wang, 2016; Gillberg等,2017; Cimmino等,2018;
Mastrangelo等,2018)。与本讨论特别相关的是2-OGDD直接参与表观遗传调控,特别是十一个11个转位酶(TET)和JMJC家族。
TET家族是在最初发现TET1作为AML中MLL与t(10; 11)(q22; q23)的易位伴侣后命名的(Ono等人,2002;
Lorsbach等人,2003)。随后在哺乳动物基因组中进行同源搜索,发现该家族的另外两个成员TET2和TET3与TET1一起催化了5-甲基胞嘧啶(5mC)向5-羟甲基胞嘧啶(5hmC)的转化(Tahiliani等,2009)。
TET蛋白还可以将5-hmC进一步氧化为5-甲酰基胞嘧啶(5fC),然后再氧化为5-羧基胞嘧啶(5caC)。这些氧化步骤共同导致DNA的主动和被动去甲基化(He等,2011;
Kohli和Zhang,2013)。
JMJC家族包含20多种蛋白质,这些蛋白质共同能够使单,二和三甲基化的组蛋白赖氨酸残基脱甲基化(Tsukada等,2006;
Monfort和Wutz,2013)。每个脱甲基步骤涉及甲基的氧化,然后自发除去甲醛。这些发现已经将TET和JMJC双加氧酶确立为分别用于DNA和组蛋白的主动去甲基化的组成部分。
在各种组织的实体瘤以及血液系统恶性肿瘤中均已检测到影响这些双加氧酶的表达或突变发生变化(Gillberg et
al。,2017)。鉴于抗坏血酸对于两个家族的最佳活性都是必需的(Klose等,2006; Tsukada等,2006;
Minor等,2013),并且这些癌症中的大多数突变都会影响该基因的一个拷贝,抗坏血酸可以通过增加剩余的酶活性来补偿。支持该机制的大多数证据来自反洗钱模型。
大型AML患者队列的下一代测序表明,约10%的患者携带TET2突变,主要仅影响一个等位基因(Ley等人,2013;
Papaemmanuil等人,2016)。
JMJC突变的发生率要低得多,约有1%的患者检测到KDM5A和KDM6A发生变化。有趣的是,IDH的突变与TET2互斥,IDH1或IDH2的突变约占20%。这种排他性的原因证明了新陈代谢与表观遗传学之间的紧密联系。
IDH将异柠檬酸转化为2-OG(TET和JmjC酶所需的辅助因子)。相比之下,突变体IDH(mutIDH)会生成2-羟基戊二酸(2-HG),已被证明可通过抑制TET2破坏造血功能(Figueroa等,2010;
Losman等,2013)。
在此信息的基础上,最近进行的三项研究探索了抗坏血酸在涉及突变TET2或IDH的小鼠和白血病细胞模型中恢复正常细胞功能的潜力(Agathocleous等人,2017;
Cimmino等人,2017; Shenoy等人) 。,2017)。 Cimmino等。
(2017)使用转基因小鼠模型,利用RNAi可诱导TET2敲低和可逆。他们表明,敲低TET2会导致AML患者自我更新异常,髓系谱系标记丢失,显着的甲基化过高以及基因甲基化降低。
TET2恢复和抗坏血酸(250μM)均可逆转这些变化。有趣的是,添加过氧化氢酶(1单位/
mL的培养基)对抗坏血酸在重新铺板试验中逆转异常自我更新的能力没有影响。此外,该浓度的抗坏血酸不会增加细胞内二氯荧光素的荧光,而细胞内活性氧的指示剂仅用50μMH2O2就可以检测到(Wang等人,2014)。
Agathocleous等。
(2017)使用了多种不同的小鼠模型,包括Tet2缺乏症,Golo-/-和Flt3ITD(与Tet2缺失协同诱导AML的激活突变)的各种组合。首先,他们发现人和小鼠HSC的抗坏血酸水平异常高,与抗坏血酸转运蛋白Slc23a2的表达水平相关。其次,他们显示Gulo-/-小鼠的HSC数量增加,部分原因是TET2活性降低。在一系列涉及将Flt3ITD供体骨髓细胞移植到野生型或Gulo-/-受体中的实验中,他们表明抗坏血酸缺乏能够与Flt3ITD协同作用,以类似于Tet2缺失的方式促进白血病的发生。此外,相对于野生型受体,当将Tet2Δ/
+; Flt3ITD供体骨髓细胞移植到Gulo-/-受体中时,加速了髓样白血病的发展。最后,在7周时向Tet2Δ/ +; Flt3ITD;
Gulo-/-小鼠补充1%的抗坏血酸饮食(Harlan),与未补充饮食的小鼠相比,显着延长了生存期。使用Tet2Δ/Δ;
Flt3ITD供体骨髓细胞进行的相关实验表明,这些作用是通过Tet2依赖性和Tet2依赖性机制介导的。
Tet2独立效应可能是通过Tet1或Tet3介导的,但这将需要在未来的实验中加以探讨。
第三项研究利用表达IDH1R132H的HOXA9永生小鼠骨髓细胞来研究抗坏血酸治疗的效果(Mingay et
al。,2018)。抗坏血酸处理(345μM)促进了与髓系分化有关的增强子的DNA脱甲基。抗坏血酸治疗还导致启动子去甲基化并增加了几个关键造血基因的表达。重要的是要注意2-磷酸抗坏血酸被用于细胞培养实验。这样做是为了明确避免细胞外产生H2O2。
累积地,这些数据表明抗坏血酸能够至少部分地通过上调TET2活性来减轻白血病模型中Tet2丢失的影响。鉴于所用的抗坏血酸浓度低,所看到的影响不太可能通过过氧化氢的产生来介导。这些发现需要对使用抗坏血酸改善TET杂合或IDH突变患者的TET活性进行调查。这与AML特别相关,其中TET2和IDH突变是早期驱动因素(Papaemmanuil等,2016)。 TET2突变发生在其他健康的成人克隆性造血患者的白细胞中(Busque等,2012),可以想象补充抗坏血酸可以预防进展为骨髓增生异常综合征或明显的AML。近期一项针对AML患者的临床试验将抗坏血酸补充剂作为60-87岁老年患者DNA甲基转移酶抑制(DNMTi)治疗的辅助手段(Zhao等人,2018)。他们比较了阿克拉比丁和阿糖胞苷之前的抗坏血酸加小剂量地西他滨(A-DCAG,n = 39)与单独使用DCAG(n = 34)。抗坏血酸组经过一轮治疗后,临床缓解率明显更高,这与更高的总生存期相关。不幸的是,没有提供关于患者组中TET2突变状态的数据。如果要在患者队列中验证表观遗传作用机制,则涉及抗坏血酸的未来临床试验将需要通过突变状态对反应进行分层。
癌症中抗坏血酸的药代动力学考虑
以上讨论的所有假设都要求通过在整个肿瘤环境中的有效分布来使抗坏血酸充分进入肿瘤细胞。了解抗坏血酸传递到细胞外肿瘤空间并被细胞吸收的药代动力学对于理解抗坏血酸的影响和设计用于临床干预研究的有效治疗方案至关重要。如上所述,口服或静脉内给药后血浆药代动力学之间存在根本差异(Padayatty等,2004)。对静脉内输注后血浆水平的模型进行建模预测,可以达到mM范围内的峰值浓度,随后在许多研究中对此进行了测量(Hoffer等人,2008;
Robitaille等人,2009;
Stephenson等人等人,2013年)。在荷瘤的Gulo-/-小鼠中,高剂量腹膜内注射后24小时内,抗坏血酸水平升高在肿瘤中持续存在,这归因于在低氧肿瘤环境中稳定性的提高(Campbell
et al。 ,2016b)。
从包括动物研究在内的所有当前证据以及可从人类临床研究中获得的信息来看,静脉内输注治疗癌症似乎确实有优势,但尚未确定剂量和给药频率的最佳方案。癌症患者通常服用约1
g /
kg的剂量(Padayatty等人,2010),并且明显具有良好的耐受性,且副作用极小(Stephenson等人,2013)。发现抗坏血酸的频率也可能很重要,这一发现表明,每天向Gulo-/-小鼠给药后,抗坏血酸的抗肿瘤活性比隔日输注时更有效(Campbell等,
2016b)。
抗坏血酸的吸收和代谢抗坏血酸是一种小的,高度水溶性的分子,来源于葡萄糖,存在于所有动植物中(Hornig,1981; Smirnoff and
Wheeler,2000; Padayatty and
Levine,2016)。大多数动物在肝脏或肾脏中合成它,但它已成为人类,其他灵长类动物,豚鼠和果蝠的维生素,它们在古龙内酯氧化酶(生物合成途径中的末端酶)中发生了突变(Chatterjee等,1975;
Banhegyi等人,1997; Linster和Van Schaftingen,2007;
Du等,2012)。在所有动物中,抗坏血酸通过循环运输到组织,大多数细胞通过钠依赖性维生素C转运蛋白SVCT1和SVCT2的主动摄取,将维生素浓缩至血浆水平(平均约50μM)的许多倍。
1997; May和Qu,2005; Savini等,2008; Harrison和May,2009; Mandl等,2009;
Nualart等,2014)。例外是红细胞,它不表达SVCT2并通过GLUT吸收脱氢抗坏血酸(DHA)来积累抗坏血酸(Tu等人,2017)。组织水平差异很大,某些器官,尤其是脑,肾上腺,肝脏和白细胞,其浓度高达20
mM(Tsao,1997)。人们认为高的细胞内水平反映了对抗坏血酸作为必需的酶辅因子的需求(Tsao,1997; May和Qu,2005)。
像所有组织细胞一样,肿瘤细胞也依赖血浆供应营养。肿瘤中血管系统不佳会阻止有效的氧气输送并导致缺氧反应的上调(Ratcliffe,2013;
Semenza,2016)。氧气在组织中的扩散距离约为100μm,这与灌注良好的正常组织中的血管间距离一致(Ratcliffe,2013;
Semenza,2016)。因此,当脉管系统功能失调和混乱时,抗坏血酸的传递可能会受到类似的损害。当在体外模拟抗坏血酸通过组织层的扩散时,发现它以细胞周围的方式扩散穿过多细胞层(Kuiper等人,2014c)。基于扩散参数和所测量的摄取到细胞中的动力学,对血浆中浓度可变的抗坏血酸在肿瘤中的可能分布进行了建模。这些数据表明,当血浆浓度非常低时,组织中几乎没有抗坏血酸,只有紧邻血管的那些细胞才能积累任何抗坏血酸(图33)。针对10μM血浆水平建模的这种情况代表了一种接近坏血病的状态。当血浆浓度为50μM(在健康范围内但仍低于血浆饱和度的水平)时(Carr和Frei,1999;
Levine等人,2001),低水平的抗坏血酸会从血管渗透到100μm,但细胞会蓄积生理低mM浓度仅在紧邻血管的细胞中可见(图图33)。当血浆饱和(100μM)时,可以达到最佳的细胞内水平,但扩散区不会超过100μm。推断100μM血浆抗坏血酸(通过口服摄入可达到的最大值)不足以确保在整个灌注不良的肿瘤中有效分配至细胞。当血浆水平增加到mM范围时,该模型表明即使在距血管200μm处也能最佳摄取细胞。该信息支持本评价中讨论的许多假设的理论基础,这些假设表明静脉抗坏血酸输注相对于口服摄入具有优势。
图3
与血浆浓度有关的抗坏血酸通过细胞外组织进入组织细胞的分布。当血浆水平低(10μM)时,实际上没有抗坏血酸能够到达组织。这表示接近坏血病(完全缺乏)的状态。当血浆水平处于“健康但不饱和”范围(50μM)或饱和(100μM)范围内时,抗坏血酸在组织细胞中积累的能力存在显着差异。低于饱和度时,只有与血管壁相邻的细胞才会积聚生理细胞内浓度(低mM)。血浆抗坏血酸≤100μM时,抗坏血酸的扩散不会超过100μm,超过此有限距离的细胞将无法积累抗坏血酸。为了达到该扩散极限,需要超生理水平。氧气在组织中的扩散距离为100μm,这是灌注良好的组织中血管之间的平均距离(Ludke等,2009,2010;
Volta等,2013)。该图中表示的数据来自Kuiper等。 (2014c)。黑条表示100μm。
摘要
最近的发现扩展了我们对抗坏血酸生物学功能的认识,并突出了许多有趣的假设,这些假设表明有很好的理由来研究将抗坏血酸用作癌症辅助治疗的可行性。我们相信,本综述中描述的研究进展将很快导致开发在癌症临床环境中使用抗坏血酸的有效方案。正在进行的临床研究将有助于确定哪些患者可能会从抗坏血酸治疗中受益。鉴于其缺乏毒性,易于获得且成本低廉,我们希望有关作用机理的良好信息将有助于将新信息转化为临床实践。
Potential Mechanisms of Action for Vitamin C in Cancer: Reviewing the
Evidence
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6037948/#B1
缺氧控制转移
Hypoxic control of metastasis
Erinn B. Rankin1,2, Amato J. Giaccia1,*
1Division of Radiation and Cancer Biology, Department of Radiation Oncology,
Stanford University Medical Center, Stanford, CA 94305-5152, USA.
2Department of Obstetrics and Gynecology, Stanford University Medical Center,
Stanford, CA 94305-5152, USA.
↵*Corresponding author. E-mail: giaccia@stanford.edu
抽象
转移性疾病是癌症相关死亡的主要原因,并且涉及肿瘤细胞与微环境之间的关键相互作用。缺氧是促进转移进展的有效微环境因子。在临床上,低氧和低氧诱导型转录因子HIF-1和HIF-2的表达与多种肿瘤类型中远处转移的增加和生存率低有关。而且,恶性细胞中的HIF信号传导影响转移级联中的多个步骤。
在这里,我们审查研究集中在阐明低氧肿瘤微环境促进转移进展的机制。这些研究已经确定了由缺氧调节的潜在生物标志物和治疗靶标,可以将其纳入旨在预防和治疗转移性疾病的策略中。
肿瘤转移是癌症临床管理中的主要挑战。转移性疾病占所有与癌症相关的死亡的90%以上,并且通常与患者高死亡率相关,因为难以通过外科手术或常规化学疗法和放射疗法进行治疗。转移是一个复杂而动态的过程,它选择具有高度侵略性的肿瘤细胞,使其具有从其原始组织中扩散,在异物组织微环境中存活并在远处生长的能力。最近的研究表明,原发肿瘤和远处组织中受微环境调节的细胞和分子成分深刻影响了肿瘤的转移倾向。 氧气是控制发育过程以及正常组织动态平衡的微环境因素。在细胞水平上,氧化代谢,生成5'-三磷酸腺苷(ATP)和细胞存活都需要氧气。为了维持氧气的稳态,后生动物利用低氧信号通路促进氧气的输送和细胞对缺氧的适应(1)。缺氧正在成为转移调控中的关键微环境因素。在这里,我们回顾我们目前对缺氧和低氧信号在转移中所起的作用的了解,并重点介绍该领域的最新进展。
缺氧信号
缺氧诱导的转录因子HIF-1和HIF-2通过激活控制葡萄糖摄取,代谢,血管生成,红细胞生成,细胞增殖,分化和凋亡的基因表达程序来协调对低氧张力的适应性细胞反应(1)。
HIF信号促进肿瘤发生的开始和发展,并且肿瘤使用多种策略激活该途径。
缺氧或低氧张力是肿瘤微环境的标志性特征。据估计,由于氧气输送和耗氧量之间的不平衡而导致的缺氧和/或缺氧区域占实体瘤的50%至60%(2)。在肿瘤微环境内,由于肿瘤脉管系统的异常,包括以毛细血管渗漏和血流缓慢为特征的扩张的毛细血管,氧气输送受到损害(3)。同时,由于肿瘤细胞增殖和免疫细胞浸润的需要,氧气消耗率很高。在临床上,缺氧与HIF活化,转移,对化学疗法和放射疗法的抵抗力以及患者生存期差有关,这表明缺氧可能有助于肿瘤进展和对治疗的抵抗力(4-6)。
缺氧通过促进HIF-α亚基的蛋白质稳定性来激活HIF信号传导。在常氧条件下,脯氨酰羟化酶(PHD 1-3,也称为EGLN
1-3)使用氧气作为底物将HIF-1和HIF-2α亚基中的关键脯氨酸残基羟化(7-9)。这种羟基化事件使E3泛素连接酶复合物的底物识别组分von
Hippel-Lindau肿瘤抑制蛋白(pVHL)与HIF-α结合并靶向其进行蛋白酶体降解(10-14)。在缺氧条件下,PHD活性受到抑制,导致HIF-α稳定并易位至细胞核。在细胞核中,HIF-α亚基与芳烃受体核转运蛋白(ARNT)二聚,并与目标基因中的缺氧响应元件结合,通过募集包括p300
/
CBP在内的转录共激活因子来激活基因转录(15-17)。数百个基因响应HIF-1和HIF-2而被激活;这些因素使细胞得以生存并适应低氧张力,并赋予两者和促进宿主转移的改变(图1)。

图1.肿瘤细胞中HIF-1和HIF-2激活的机制。
缺氧是癌症中HIF激活的常见机制。在常氧条件下,PHD酶(也称为EGLN
1-3)利用氧气作为底物将位于HIF-α亚基内的关键脯氨酸残基羟化。该羟基化事件介导pVHL结合以及随后的26S蛋白酶体的泛素化和降解。在缺氧或pVHL丧失的情况下,HIF-α稳定并易位至细胞核,在该细胞中与ARNT异源二聚体,并与靶基因调控区域内的缺氧反应元件(HRE)结合。当辅因子(p300
/ CBP)募集时,HIF异二聚体激活这些位点的基因表达。
PRC2介导的组蛋白甲基化可以抑制参与转移的HIF目标基因的激活。还可以通过增强HIF-α产生(TORC1,YB-1)或防止其降解(UCHL1,WSB1)的机制在肿瘤细胞中诱导HIF活性。
除了通过缺氧激活外,PHD-VHL-HIF信号通路还可以通过遗传事件和其他微环境因素来调节。 EGLN
1-3的酶活性需要铁和2-氧戊二酸酯来使HIF-α亚基羟基化。结果,EGLN
1-3的活性和HIF-α的稳定性受铁的有效性影响,并且可以被Krebs循环中间体(包括琥珀酸酯和富马酸酯)与2-氧戊二酸酯竞争而被抑制(18-22)。在肿瘤细胞中,琥珀酸脱氢酶(SDH)或富马酸水合酶(FH)编码基因的突变会导致琥珀酸或富马酸胞质的积累。这抑制了脯氨酰羟化酶的活性,并在常氧条件下导致HIF稳定[有关最新综述,请参见(22)]。此外,线粒体产生的活性氧(ROS)可以抑制PHD活性。然而,在缺氧情况下发生这种现象的机制仍未确定(图1)(21、23)。
如上所述,VHL通过控制HIF-α亚基的泛素化和降解来调节低氧信号传导。 VHL的基因突变或表观遗传失活会导致VHL疾病,这与HIF-1和HIF-2的组成性激活以及高度血管化的肿瘤(包括血管母细胞瘤,嗜铬细胞瘤,胰岛细胞瘤和透明细胞肾细胞癌)的发展有关( ccRCC)。肾细胞癌是VHL患者发病的主要原因,因为这些肿瘤具有转移到其他组织部位的能力(24)。此外,大多数零星的ccRCC会通过基因组改变而失去VHL功能(25)。尽管VHL的基因失活被认为是ccRCC发病机制中的早期事件,但最近的工作已经确定了肿瘤进一步增强HIF活性以促进转移进程的机制。通过分析VHL缺陷型ccRCC细胞的转移亚群,Vanharanta及其同事发现,前转移基因内的表观遗传修饰可以增强与转移相关的HIF靶基因的表达(26)。例如,依赖于多梳抑制复合物2(PRC2)的组蛋白H3 Lys-27三甲基化的丧失激活了HIF介导的趋化因子(C-X-C基序)受体4(CXCR4)的表达,从而促进了侵袭和转移(26)。这些发现打开了新的研究领域,旨在了解在肿瘤进展过程中前转移基因位点的表观遗传学变化是如何改变的。最近的研究还表明,肿瘤中的VHL活性可以由WSB1调控,WSB1是E3连接酶,靶向pVHL进行泛素化和蛋白酶体降解(27)。 WSB1在肿瘤细胞中的表达在常氧和低氧条件下导致HIF稳定,并与癌症患者的转移相关(图1)(27)。
最后,缺氧信号传导可以通过促进HIF-α产生或阻止其降解的因子来激活。 HIF-αmRNA的转录和翻译均受TORC1活性的调节[综述于(22)]。磷酸酶和张力蛋白同源物(PTEN)或结核硬化复合物(TSC)复合物中的失活突变以及磷脂酰肌醇3-激酶(PI3K)或AKT中的活化突变导致TORC1活性增加,从而促进了HIF-α的稳定(22, 28-31)。另外,通过生长因子受体信号传导激活PI3K和AKT信号可以促进常氧条件下HIF-α的稳定和活性(32,33)。 HIF-1 mRNA的翻译可以在常氧和低氧条件下通过结合YB-1,一种RNA和DNA结合蛋白来增强。这会导致肉瘤细胞中的HIF-1蛋白增加和转移活性增强(34)。还可以通过上调阻止HIF-α降解的因子来促进HIF活性。 Goto等人使用一种优雅的筛选方法来识别在常氧条件下会导致HIF-1转录活性的因素。最近,泛素C末端水解酶L1(UCHL1)被鉴定为HIF-1去泛素化酶,可通过防止VHL介导的HIF-1降解来促进在常氧和低氧条件下的HIF-1活性(图1)(35)。 UCHL1与HIF-1和癌症患者的远处转移有关,提示UCHL1可能通过HIF促进转移(35)。为了支持这一概念,UCHL1过表达细胞中HIF-1的基因失活逆转了这些细胞的转移潜能(35)。总之,这些研究强调了在缺氧和常氧条件下可以激活HIF信号的多种机制,并且可能在转移过程中促进了HIF的激活(图1)。
在临床上,HIF-1和HIF-2在原发肿瘤和转移灶中高表达。对人类原发性肿瘤样本的免疫组织化学分析显示,HIF-1高表达与妇科,胰腺癌,食管癌,肺癌和前列腺癌患者的转移相关(36-38)。
HIF-2在原发性肿瘤中的表达还与小细胞肺癌和乳腺癌患者的远处转移有关(39、40)。而且,在多种人类癌症中,HIF表达增加通常与患者死亡率增加相关(41)。在实验模型中,HIF在肿瘤细胞中的过表达促进转移(42、43),而HIF失活则降低了肿瘤细胞的转移潜能(43-47)。总之,这些临床和实验结果证明了HIF信号在转移性肿瘤进展中的重要作用。然而,在将这些知识转化为安全有效的抗转移疗法之前,至关重要的是要了解癌细胞和基质细胞如何利用该途径来支持转移。
HIF介导的转移机制
缺氧和HIF信号的激活影响转移级联反应的多个步骤,包括侵袭和迁移,血管内和外渗,转移前生态位的建立以及远处的存活和生长[图10]。
2;有关最新评论,请参见(48,49)]。在这里,我们重点介绍了最近的研究,这些研究揭示了HIF信号促进转移进展的新机制。
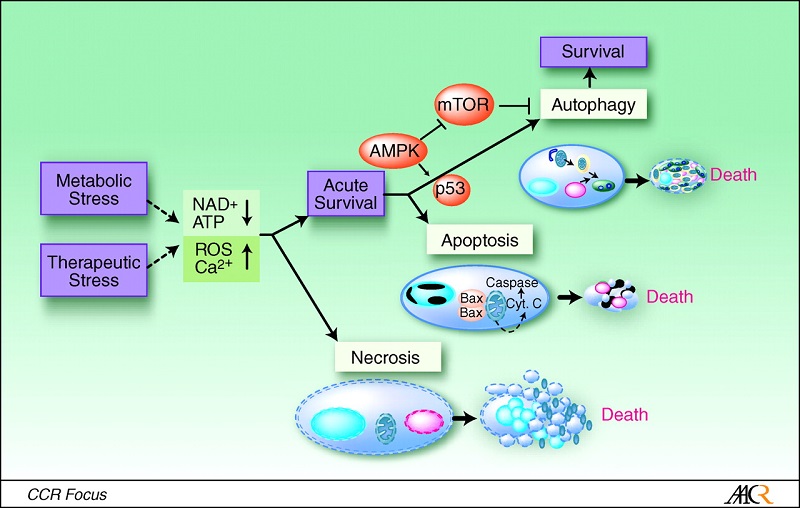
图2 HIF信号调节转移级联内的多个步骤。
括号中突出显示了HIF的直接靶基因,可促进转移的每个步骤(在所示示例中为肺)。 ECM,细胞外基质; BMDC,骨髓来源的细胞。
缺氧信号和免疫逃避
转移性肿瘤进展中的关键步骤是肿瘤细胞逃避免疫攻击的能力。肿瘤缺氧被认为促进肿瘤细胞和浸润性免疫细胞的免疫抑制表型。在临床前模型中,呼吸性高氧(60%O2)可以促进肿瘤消退,减少转移性疾病并延长动物存活(50)。在这些研究中,呼吸性高氧症与肿瘤内缺氧的减少和以大量CD8
+
T细胞浸润和调节性T细胞浸润减少为特征的免疫允许的肿瘤微环境有关(50)。了解缺氧促进免疫抑制的机制是一个活跃的研究领域,并且在转移性疾病的治疗中可能具有重要的治疗意义。
缺氧通过几种HIF依赖性机制促进肿瘤对免疫攻击的抵抗力。
HIF信号在肿瘤细胞中的常氧和低氧稳定作用增强了对细胞毒性T淋巴细胞介导的裂解的抵抗力(51-54)。缺氧和HIF介导的肿瘤细胞自噬激活也调节自然杀伤(NK)细胞介导的抗肿瘤反应。这是由于自噬小体中NK衍生的颗粒酶B的低氧降解和1型肌醇1,4,5-三磷酸受体的诱导而引起的(55,56)。缺氧信号传导可通过增加肿瘤细胞和髓样抑制细胞(MDSC)上程序性死亡配体1(PD-L1)的表达以及增强CD8
+ T细胞上CTLA-4的表达来增强对T细胞介导的杀伤的抗性。
,分别[在(57)中进行了审查]。此外,低氧肿瘤细胞通过上调CD47来逃避先天免疫识别,CD47是一种细胞表面分子,可与巨噬细胞表面的信号调节蛋白α(SIRPα)相互作用,从而阻止吞噬作用(58)。
缺氧和HIF通过将调节性T细胞(Treg),MDSC和巨噬细胞募集到肿瘤微环境中来促进免疫抑制性微环境[综述(57,59)]。缺氧的肿瘤细胞通过分泌CCL28,转化生长因子-β(TGF-β)和血管内皮生长因子[10],将表达CCR10(CC趋化因子受体10型)和NP-1(神经纤维蛋白-1)的Treg募集到肿瘤微环境中[
VEGF;
(57)所述]。一旦进入肿瘤微环境,Treg就会促进免疫耐受和血管生成,从而支持转移性肿瘤的生长(60)。同样,低氧肿瘤细胞通过分泌趋化因子和细胞因子,包括CCL5,C-X-C基序趋化因子12(CXCL12或SDF-1),VEGF和内皮素[ET-1和ET-2;
(57)所述]。
MDSC和巨噬细胞中的低氧信号通过促进免疫抑制表型和刺激血管生成而直接促进肿瘤进展[综述于(57,61–63)]。此外,低氧主要通过细胞外腺苷的积累来抑制肿瘤浸润淋巴细胞的效应子功能。腺苷通过T细胞上的A2A受体发出信号,从而增加环状腺苷3',5'-单磷酸,从而抑制T细胞增殖,扩增和细胞因子分泌[综述于(59)]。总的来说,这些发现说明了肿瘤细胞,MDSC和肿瘤相关巨噬细胞内HIF信号转导有助于建立免疫抑制性肿瘤微环境的多种方式。
EMT,入侵和迁移中的HIF信号
在转移的早期阶段,肿瘤细胞具有侵袭和迁移特性,使它们能够离开局部原发肿瘤块并进入邻近和远处的宿主组织。肿瘤细胞利用多种迁移和侵袭机制,包括个体和集体细胞迁移策略(64)。体外肿瘤细胞迁移与上皮-间质转化(EMT)相关。
EMT的特征在于细胞形态的变化,以及细胞与细胞和细胞与基质的粘附。细胞间粘附主要是由在细胞间连接处表达的钙黏着蛋白介导的(65)。
E-钙黏着蛋白的丢失使细胞脱离其邻居,并开始向循环系统或淋巴系统迁移,以寻找新的地形。
E-钙粘蛋白的表达通常在转移性肿瘤中观察到,并且与实验模型一起工作证明,E-钙粘蛋白的丧失足以促进恶性细胞的转移(66)。
HIF的缺氧或过表达足以诱导多种细胞类型的EMT和侵袭(42,67–71)。 HIF信号通过两种直接和间接机制来促进EMT。 HIF在关键EMT转录因子(如ZEB1,Snail和Twist)的调控中具有直接作用,这已经通过在这些基因的调控元件内鉴定功能性缺氧反应元件(HRE)得以证实(图2)(42, 72,73)。 HIF途径还通过许多细胞信号途径间接促进EMT,包括Notch(74,75),TGF-β(76),整联蛋白连接的激酶(77),某些酪氨酸激酶受体(48),Wnt(73)和刺猬(78,79)。我们最近发现,AXL受体酪氨酸激酶是一种新型的HIF靶标,可驱动VHL缺乏和低氧的癌细胞中的EMT,侵袭和转移(80)。越来越多的文献支持AXL在促进多种肿瘤类型(包括乳腺(81),卵巢(82)和肺(83))内转移的重要作用。此外,据报道,AXL抑制可使耐药的间充质肿瘤细胞对抗癌药敏感,包括抗有丝分裂药,表皮生长因子受体(EGFR)抑制剂和PI3K抑制剂(83-86)。 AXL的丧失似乎对小鼠的胚胎发育或正常的成年组织功能没有有害作用(87、88)。此外,AXL的生物抑制剂(例如可溶性AXL诱饵受体)具有特异性,并且与小鼠的正常组织或血液学毒性无关(82、89)。因此,AXL似乎是治疗晚期癌症的有希望的治疗靶标,并且目前正在临床前和临床开发中使用多种AXL信号抑制剂。
HIF信号传导和转移前的环境
转移的晚期由散布的肿瘤细胞在远处的组织微环境中定植,存活和生长的能力决定。在过去的十年中,小鼠实验令人信服地表明,在次级器官中形成促进肿瘤的转移前生态位对于在远处转移和生长的转移性肿瘤细胞至关重要(90)。转移前利基部分地由肿瘤分泌的因子建立的发现引起了人们对确定支配组织特异性转移的因子进行潜在治疗靶向的兴趣。肿瘤微环境因素如缺氧在调节这些分泌因子中的作用同样是一个深入研究的领域。
原发肿瘤中的HIF信号有助于促成转移前生态位形成的分泌因子的产生(图2)。在乳腺癌细胞中,HIF信号传导导致赖氨酰氧化酶(LOX)和LOX样蛋白(LOXL2和4)的表达和分泌增加。这些蛋白质修饰肺中的胶原蛋白基质,以募集骨髓来源的细胞(BMDC),这些细胞可引发肺部转移性定居。
BMDC通过产生趋化因子促进转移,这些趋化因子通过刺激肿瘤细胞外渗的机制将肿瘤细胞募集到肺中(44、90-95)。最近的研究表明,LOX也是在骨中建立转移前利基的关键因素。与MDA-231亲代细胞相比,在骨性MDA-231人乳腺癌细胞中低氧分泌组的蛋白质组学分析确定LOX是在骨性细胞中分泌量最高的分泌蛋白之一(96)。此外,LOX表达在临床上与雌激素受体阴性(ER–)乳腺癌转移至骨有关。在临床前模型中,即使在肿瘤细胞到达之前,原发性肿瘤内的MDA-231骨性细胞中的LOX表达对于诱导免疫受损小鼠的溶骨性病变和皮质骨丢失都是必要且充分的。
LOX调节破骨细胞的生成,发现LOX在原发性肿瘤中的高表达与小鼠溶骨性骨病变的形成有关(96)。这些发现表明,LOX可能是鉴定有骨复发风险的ER阴性患者的重要生物标志物。它们还增加了抑制LOX可能预防乳腺癌患者骨和肺复发的可能性。鉴于LOX还可以促进肿瘤细胞的增殖,侵袭和血管生成,因此LOX是癌症治疗的有吸引力的治疗靶标(97)。
乳腺癌细胞调节转移前生态位的另一种机制是通过肿瘤-淋巴管串扰。远处的淋巴管支持乳腺癌,黑色素瘤,前列腺癌,胃癌和结肠癌患者的转移性定植(98)。淋巴管内皮细胞(LECs)是一种专门的内皮细胞,位于淋巴管内并通过主动将肿瘤细胞募集到淋巴管而促进淋巴转移。
LEC通过产生趋化因子(包括SDF-1和CCL21)来实现这一目标,所述趋化因子分别与肿瘤细胞表达的同源受体CXCR4和CCR7结合(99)。转移前器官内的LEC受肿瘤衍生因子的调节,以促进肿瘤细胞募集,外渗和定植(98)。肿瘤分泌的IL-6激活位于淋巴结和肺内的LEC中的信号转导子和转录3(STAT3)信号激活子,以促进(i)CCL5介导的CCR5阳性乳腺癌细胞募集入淋巴系统,和(
ii)VEGF介导的肺血管通透性和淋巴结血管生成(98)。有趣的是,IL-6诱导的LECs中的VEGF表达与HIF-1的激活有关,表明肿瘤来源的因子可能至少部分通过LECs中HIF信号的激活促进淋巴转移(98)。总的来说,这些研究表明,缺氧和HIF信号通过激活循环因子的表达来介导肿瘤基质间的串扰,所述循环因子可以引发和指导转移性生长。
令人关注的新兴领域是外泌体在转移中的作用以及低氧和HIF信号传导是否可能通过这种机制促进转移前利基的形成。外泌体是通过携带和转移包括蛋白质,脂质,微小RNA和mRNA在内的分子来调节细胞间通讯的细胞外小泡(100)。外来体最近与转移前利基的建立有关。它们以高水平存在于癌症患者的血清中,并与晚期疾病有关。此外,肿瘤来源的外来体优先定位于转移部位,包括肝,肺和骨髓,在这些部位它们可促进血管通透性并增加BMDC的募集(101-104)。但是,原发肿瘤中调节外泌体含量和释放的因素仍然未知。最近的研究表明,外泌体通过激活内皮细胞和周细胞中旁分泌信号传导来介导多形性胶质母细胞瘤(GBM)中的血管生成的缺氧依赖性刺激(105)。有趣的是,从GBM患者血浆中分离出的外泌体包含的缺氧调节蛋白数量有所增加,包括MMP9,MMP8,血小板衍生生长因子(PDGF),PAI1和胰岛素样生长因子结合蛋白3(IGFBP3)。从性和年龄匹配的对照组血浆中分离出来的外泌体(105)。这项工作表明缺氧可以影响外泌体货物的含量以促进血管生成。此外,这些发现表明,外泌体的低氧信号可以作为预测恶性肿瘤侵袭性的非侵入性生物标记。除调节外泌体货物含量外,缺氧还促进微囊泡脱落。缺氧细胞以HIF依赖性方式上调小鸟苷三磷酸酶RAB22A,以促进微囊泡脱落,侵袭和转移(106)。
HIF信号传导以及远处细胞的生长和存活
成功的转移定植需要扩散的肿瘤细胞适应远处组织的微环境,这可能与携带原发肿瘤的组织的微环境非常不同。为了说明这一点,我们讨论了肝脏,这是结肠直肠癌的常见转移部位。肝脏通过控制碳水化合物和脂质代谢来维持总体能量平衡。在肝脏内,建立了一个氧气梯度,为器官内的代谢活动提供了边界(图3)。门静脉血中的氧分压为60至65
mm Hg,在静脉血中为30至35 mm
Hg(107)。因为氧气是氧化代谢的必需电子受体,所以执行葡萄糖或脂肪酸氧化的肝细胞位于有氧的门静脉区,而与氧气无关的代谢功能(例如葡萄糖摄取,糖酵解和脂肪酸合成)主要由静脉肝细胞执行(108)。鉴于结肠癌细胞通过门脉循环转移到肝脏,Loo及其同事(109)假设这些细胞会经历急性缺氧并竞争糖酵解底物。
他们使用HIF-1转录荧光素酶报告基因小鼠确定了结肠癌细胞在肝扩散后早期就处于缺氧状态。他们发现,已扩散的癌细胞增加了肌酸激酶脑型酶(CKB)的释放,这有助于它们在肝脏缺氧的微环境中生存(109)。
CKB通过催化高能磷酸基团从ATP到代谢物肌酸的转移,产生磷酸肌酸来控制快速动员的高能磷酸根的量(110)。在诸如缺氧等代谢应激条件下,肿瘤细胞利用磷酸肌酸储备作为高能磷酸盐的来源,可将其转移至5'-二磷酸腺苷(ADP)生成ATP(110)。
CKB的耗竭增加了肝脏缺氧肿瘤细胞中caspase介导的细胞死亡,这表明肝低氧是结肠癌细胞在转移性传播早期的障碍,并且细胞通过从磷酸肌酸储备中生成ATP克服了这种代谢压力(图3)。
(109)。

图3肝转移中代谢重编程的机制。
肝脏微环境缺氧。吡莫硝唑染色(棕色)表明肝脏缺氧区域(中图)。转移性肿瘤细胞进入肝脏缺氧区域,必须代谢适应以抵抗与缺氧相关的代谢应激。散布的乳腺癌细胞(右)通过增加丙酮酸脱氢酶激酶1(PDK1)的表达并促进糖酵解的重新编程来适应新陈代谢。扩散的结肠癌细胞(左)通过增加肌酸激酶脑型(CKB)的表达和分泌来代谢适应。该酶通过催化高能磷酸基团从ATP到代谢物肌酸的转移,从而产生磷酸肌酸来控制快速动员的高能磷酸根的量。在诸如缺氧等代谢应激条件下,肿瘤细胞利用磷酸肌酸储备作为高能磷酸盐的来源,可将其转移至ADP以生成ATP。
Dupuy及其同事观察到肝脏缺氧及肝周围区域相关的糖酵解表型为转移性定居提供了障碍,这一观点与这一概念一致.Dupuy及其同事观察到,与骨骼和肺转移相比,乳腺转移至肝脏的生存率高度依赖于糖酵解(
111)。肝转移性乳腺癌细胞的代谢重编程很大程度上取决于HIF-1目标基因丙酮酸脱氢酶激酶(PDK1)。
PDK1通过拮抗丙酮酸脱氢酶(PDH)的功能来抑制线粒体功能,丙酮酸脱氢酶是丙酮酸转化为乙酰辅酶A并进入三羧酸循环的限速酶(112)。值得注意的是,与原发肿瘤标本相比,PDK1表达在乳腺癌肝转移中升高,这进一步支持了PDK1表达对于肝微环境中乳腺癌细胞的生长和存活特别重要的观点。这些发现表明,乳腺癌细胞通过激活HIF信号和PDK1介导的糖酵解重编程来适应缺氧性肝微环境(图3)。因此,这些研究表明,肝脏微环境中的缺氧选择了具有代谢能力适应低氧应激的弥散性肿瘤细胞。
最后,HIF通过刺激血管生成促进远处转移的晚期。像原发肿瘤一样,转移也需要血管生成来支持其生长。
VEGF-A是肿瘤细胞产生的促血管生成因子,可刺激内皮细胞的募集和增殖,并促进周细胞的增殖和迁移(113)。
VEGF-A是一个公认的HIF靶标,它的表达是由HIF信号在原发性肿瘤和转移中诱导的(114)。
结论
缺氧和依赖HIF的信号在转移性肿瘤进展中起重要作用。尽管缺氧是激活肿瘤内HIF信号的重要因素,但最近的研究表明,恶性细胞通过促进HIF-αmRNA的翻译,蛋白质稳定性和下游靶基因表达,在发展过程中使用多种策略来增强HIF信号。
缺氧肿瘤微环境影响转移的早期和晚期。低氧应激通过促进免疫抑制和肿瘤抗性在免疫逃逸中起重要作用。在原发性肿瘤内,HIF依赖性基因表达控制EMT,侵袭,迁移和血管生成,以支持转移的早期阶段。最近的工作已确定受体酪氨酸激酶AXL是依赖HIF的侵袭和转移的关键介质,并且是预防和治疗转移性疾病的潜在治疗靶标。另外,HIF信号传导促进分泌因子如LOX,LOX样蛋白和外泌体的产生,从而在乳腺癌患者的肺和骨骼内建立了转移前环境。远处的缺氧在选择可以抵抗与缺氧有关的代谢应激的转移性细胞中也起着重要作用。转移性乳腺癌细胞在肝脏缺氧环境中激活HIF信号,以代谢适应糖酵解环境。此外,结肠癌细胞通过将CKB释放到细胞外空间中以在该微环境中生成和导入磷酸肌酸作为ATP生成的来源,从而适应缺氧肝脏内的代谢压力。
总而言之,最近的研究已经开始强调缺氧和HIF依赖性信号传导促进转移性肿瘤进展的各种机制。结果,已经鉴定了许多新的生物标志物和治疗靶标,它们对于转移性疾病的检测和治疗可能具有潜在的价值。
Hypoxic control of metastasis
Erinn B. Rankin1,2, Amato J. Giaccia1,*
Abstract
Metastatic disease is the leading cause of cancer-related deaths and involves
critical interactions between tumor cells and the microenvironment. Hypoxia is a
potent microenvironmental factor promoting metastatic progression. Clinically,
hypoxia and the expression of the hypoxia-inducible transcription factors HIF-1
and HIF-2 are associated with increased distant metastasis and poor survival in
a variety of tumor types. Moreover, HIF signaling in malignant cells influences
multiple steps within the metastatic cascade. Here we review research focused on
elucidating the mechanisms by which the hypoxic tumor microenvironment promotes
metastatic progression. These studies have identified potential biomarkers and
therapeutic targets regulated by hypoxia that could be incorporated into
strategies aimed at preventing and treating metastatic disease.
Tumor metastasis is a major challenge in the clinical management of cancer.
Metastatic disease is responsible for more than 90% of all cancer-related deaths
and is often associated with high patient mortality because it is difficult to
treat surgically or with conventional chemotherapy and radiation therapy.
Metastasis is a complex and dynamic process that selects for highly aggressive
tumor cells that acquire the ability to disseminate from their tissue of origin,
survive within foreign tissue microenvironments, and grow at distant sites.
Recent studies have demonstrated that cellular and molecular constituents
regulated by the microenvironment in both the primary tumor and distant tissue
profoundly influence the propensity of tumors to metastasize.
Oxygen is a microenvironmental factor that controls developmental processes, as
well as normal tissue homeostasis. At the cellular level, oxygen is required for
oxidative metabolism, the generation of adenosine 5′-triphosphate (ATP), and
cell survival. To maintain oxygen homeostasis, metazoan organisms use the
hypoxic signaling pathway to facilitate oxygen delivery and cellular adaptation
to oxygen deprivation (1). Hypoxia is emerging as a key microenvironmental
factor in the regulation of metastasis. Here we review our current knowledge of
the roles that hypoxia and hypoxic signaling play in metastasis and highlight
recent advances within this field.
Hypoxic control of metastasis | Science
https://science.sciencemag.org/content/352/6282/175.full
Extracellular Metabolic Energetics Can Promote Cancer ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4312495
Jan 29, 2015 · Phosphocreatine is then imported into disseminated colorectal
cancer cells where its high-energy phosphate is used to generate intracellular
ATP that sustains the energetic requirements of colon cancer cells encountering
hepatic hypoxia, allowing them …
Cited by: 161
Publish Year: 2015
Author: Jia Min Loo, Alexis Scherl, Alexander Nguyen,
细胞外代谢能可促进癌症进展
摘要
大肠癌主要转移到肝脏,每年杀死60万人。通过在肝定植过程中并行平行筛选661个miRNA,我们已经鉴定出miR-551a和miR-483是肝定植和转移的强大内源性抑制剂。这些miRNA会聚地靶向脑型肌酸激酶(CKB),该酶使代谢物肌酸磷酸化,从而产生磷酸肌酸。
CKB由遇到肝缺氧的转移细胞释放到细胞外空间,并催化细胞外磷酸肌酸的产生,磷酸肌酸通过SLC6A8转运蛋白输入并用于产生ATP,从而促进转移生存。通过单剂量腺相关病毒(AAV)联合递送miR-551a和miR-483-5p的组合治疗性病毒递送显着抑制了结肠癌转移性定植,使用小分子抑制剂抑制了CKB。重要的是,相对于原发性肿瘤,人肝转移灶的CKB和SLC6A8水平较高,而miR-551a
/ miR-483水平较低。我们确定细胞外空间为恶性能量催化和治疗靶向的重要隔室。
NF-κB对缺氧诱导因子-1α的调控
Regulation of hypoxia-inducible factor-1α by NF-κB
College of Life Sciences, Wellcome Trust Centre for Gene Regulation and
Expression, MSI/WTB/JBC Complex, Dow Street, University of Dundee, Dundee DD1
5EH, Scotland, U.K.
摘要
HIF(缺氧诱导因子)是低氧张力激活的主要转录因子。
HIF-1α(和其他α亚基)主要通过一类称为PHD(脯氨酰羟化酶)1、2和3的酶的协同作用而在蛋白质水平上受到严格控制。HIF的大多数知识来自缺氧后的研究强调;然而,在非低氧条件下也通过未知机制发现了HIF-1α的稳定作用。在本研究中,我们证明了NF-κB(核因子κB)是HIF-1α表达的直接调节剂。
HIF-1α启动子对选择性NF-κB亚基有反应。针对单个NF-κB成员的siRNA(小干扰RNA)研究显示,其对HIF-1αmRNA水平的影响不同,这表明NF-κB可以调节基础HIF-1α的表达。最后,当通过TNFα(肿瘤坏死因子α)治疗诱导内源性NF-κB时,HIF-1α水平也以NF-κB依赖性方式改变。总之,我们发现NF-κB可以调节基础TNFα,在某些情况下还可以调节低氧诱导的HIF-1α。
关键词:缺氧,缺氧诱导因子1(HIF-1),核因子κB(NF-κB),肿瘤坏死因子(TNF)
缩写:ChIP,染色质免疫沉淀; EMSA,电泳迁移率变动分析; GLUT,葡萄糖转运蛋白; HEK,人类胚胎肾脏; HIF,缺氧诱导因子;
IκB,核因子κB抑制剂; IKK,IκB激酶; NF-κB,核因子κB; PCNA,增殖细胞核抗原; PHD,脯氨酰羟化酶;
qRT-PCR,定量逆转录酶PCR; ROS,活性氧; RT,逆转录酶; siRNA,小干扰RNA; TNFα,肿瘤坏死因子α; VEGF,血管内皮生长因子
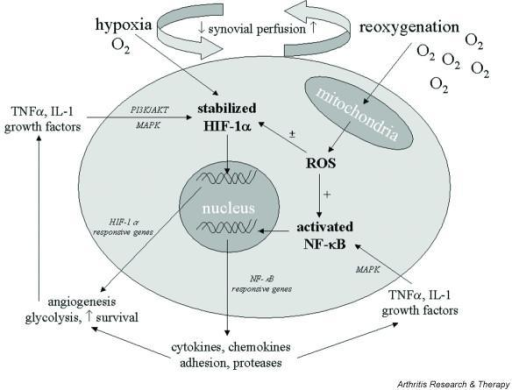
介绍
HIF(缺氧诱导因子)在发育,生理过程和病理状况中起着关键作用,因为它的存在会影响生存,细胞周期进程和代谢[1,2]。尽管HIF首先被确定为在低氧张力下激活的主要转录因子,但在常氧条件下,HIF是被细胞因子,癌基因和ROS(活性氧)激活的关键转录因子[3]。
HIF是由α和β亚基组成的异二聚因子[也称为ARNT(芳烃核转运子)]
[1]。尽管HIF-1β组成型表达且不受氧水平的调节,但是HIF-α亚基在蛋白水平上受到严格控制。这种现象主要是通过一类称为PHD(脯氨酰羟化酶)的酶的协同作用而发生的,该酶催化HIF-α亚基的脯氨酰羟化,从而不断地将它们靶向VHL(von
Hippel–Lindau蛋白)依赖性的26S蛋白酶体降解[4]。 ]。 PHD酶需要分子O2、2-氧代戊二酸酯,铁离子(Fe2
+)和抗坏血酸才能充分发挥活性,并在低氧条件下被抑制[4]。
到目前为止,已经确定了四个PHD,但是就HIF调制而言,只有三个PHD 1、2和3在功能上得到了表征[4]。缺氧后,PHD2被认为是控制HIF-1α的主要PHD
[5]。此外,所有三个PHD均可通过HIF和氧气水平进行调节[6-8]。
除了受脯氨酰羟化作用控制的HIF-α稳定机制外,HIF-1α中的Asn803或HIF-2α中的Asn851的羟化作用还可以防止与组蛋白乙酰转移酶{p300
/ CBP [CREB(cAMP-response-element-结合蛋白]-结合蛋白]},它们起着转录共激活子的作用。这种修饰改变了HIF-1 /
HIF-2激活其靶基因的能力[9]。尽管有关HIF-1的大多数知识都来自低氧应激后的研究,但在非低氧环境(例如相对充氧的肿瘤区域)以及风湿性关节炎和糖尿病等疾病中也发现了HIF-1的稳定[
10,11]。但是,尚未阐明在这些条件下实现HIF稳定的机制。已知许多在常氧中诱导HIF-1的刺激会激活许多其他转录因子,例如NF-κB(核因子κB)。因此,有可能在这两个转录因子之间发生串扰。此外,HIF-1α和PHD1启动子包含NF-κB结合位点,尚未在功能上进行表征。重要的是,在常氧条件下,HIF-1α的稳定和通过H2O2的诱导被证明是NF-κB依赖性的[12]。
NF-κB是转录因子的统称,以异二聚体或同二聚体形式存在,由称为RelA(p65),RelB,cRel,p50及其前体p105(NF-κB1)的亚基家族形成,以及p52及其前体p100(NF-κB2)。一些二聚体比其他二聚体更为普遍,它们在细胞质和细胞核之间穿梭,但主要以非活性状态被螯合在细胞质中,并被IκB(NF-κB抑制剂)抑制[13]。受到刺激后,通过诸如TNFα(肿瘤坏死因子α),癌基因或紫外线等化合物,激酶信号级联反应会导致IκB磷酸化,信号化泛素介导的蛋白酶体降解,从而导致NF-κB释放并易位至核内[13]。
,14]。然后,活化的NF-κB二聚体与细胞核中的靶DNA序列结合,从而调节NF-κB靶基因的转录。尽管NF-κB的活化非常快,但NF-κB活性的循环性质,与翻译后修饰以及靶基因上的乙酰基过高和乙酰基过低相结合,表明监管复杂性进一步提高[15]。
已经有几项研究证明了NF-κB和HIF信号通路之间存在相互干扰,包括共有的靶基因,但直接的联系尚待阐明。在本研究中,我们显示了几个NF-κB亚基与HIF-1α启动子结合。 NF-κB消耗导致基础水平的HIF-1αmRNA降低。此外,我们证明了TNFα诱导的NF-κB可以增加HIF-1αmRNA,蛋白质和活性水平,从而导致正常氧中靶基因的反式激活。仅当NF-κB通路被完全阻断时,才能观察到缺氧后HIF-1α水平的变化。这些结果表明,NF-κB可以直接调节HIF-1α途径,并且这种调节足以改变体内HIF靶基因的表达。
结果
NF-κB亚基对HIF-1α启动子的差异控制
HIF-1α启动子最初于1996年克隆[18],Michiels及其同事进行的一项研究[19]确定了转录因子如AP-1(激活蛋白1)和NF-κB的几个结合位点[19]。
。鉴于我们对NF-κB共有位点的了解不断提高[14],我们重新分析了HIF-1α启动子(http://www.genomatix.de/products/MatInspector)。该分析确定了一个距起始位点-197/188
bp的假定的NF-κB结合位点(图1a)。使用带有或不带有假定的NF-κB结合位点的HIF-1α启动子报告基因构建体[19],我们研究了正常氧血症中单个NF-κB亚基的贡献(图1a和1b).1b)。所有NF-κB亚基均可以激活HIF-1α启动子,p50的效果最高,而p52的效果最低。相反,没有NF-κB亚基激活没有NF-κB位点的HIF-1α启动子构建体的截短形式(图1b)。为了研究NF-κB是否可以结合HIF-1α启动子上的推定位点,我们使用了HEK-293细胞核提取物对NF-κB进行了EMSA提取,该细胞已被单个NF-κB亚基转染(图1c)。我们可以通过p50检测到强烈的结合,而通过p65,c-Rel和p52检测到较弱的结合。尽管表达水平相当(图1e),但用RelB不能观察到可见的结合。与HIV启动子中的经典位点结合用于评估NF-κB结合能力(图1d)。这些结果表明,NF-κB能够调节HIF-1α启动子活性。
包含图片,插图等的外部文件。
对象名称是bic874i001.jpg
在单独的窗口中打开
图1
NF-κB亚基可激活HIF-1α启动子
(a)HIF-1α启动子-荧光素酶构建体的示意图。
(b)将HEK-293细胞与1μgHIF-1α启动子-荧光素酶构建体以及1μg每种NF-κB亚单位共转染。转染后48小时测量荧光素酶活性。值是平均值+标准差。至少要进行一式三份的独立实验。
y轴显示高于对照质粒的倍数活化。
(c)NF-κB亚基与HIF-1α启动子中的κB位点结合。用每种NF-κB亚基2μg转染HEK-293细胞,制备核提取物,并使用HIF-1α启动子特异性探针通过EMSA测量DNA结合活性。
(d)如(c)中所述制备核提取物,并使用经典的NF-κB靶标启动子HIV测量DNA结合活性。 (e)通过蛋白质印迹分析核提取物中各个NF-κB亚基。
NF-κB调节基础HIF-1αmRNA水平
为了研究内源性NF-κB是否可以调节HIF-1α启动子,使用了针对不同NF-κB亚基的siRNA寡核苷酸。 siRNA序列先前已得到验证[16,17]。
qRT-PCR分析表明内源性NF-κB调节了基础HIF-1αmRNA水平(图2a)。有趣的是,p50水平的降低对HIF-1αmRNA水平没有影响,可能表明p52或其他NF-κB亚基具有补偿作用。在基因敲除小鼠中,最好地描绘了p50和p52之间的合作,其中p50和p52的单个缺失没有显示发育缺陷,而这些基因的两次缺失导致骨骼发育严重受损[20,21]。内源性p65,RelB和p52对HIF-1αmRNA水平的影响最大(图2a)。尽管诱导了HIF-1αmRNA的轻微降低,但c-Rel耗竭并未引起统计学上的显着影响(图2a)。为了评估NF-κB是否直接调节HIF-1αmRNA水平,进行了ChIP分析。在HIF-1α启动子上可以找到几个NF-κB亚基。但是,我们无法在该基因的控制区域检测到任何NF-κB结合(图2b)。这表明NF-κB直接调节HIF-1αmRNA的基础水平。
包含图片,插图等的外部文件。
对象名称是bic874i002.jpg
图2
内源性NF-κB亚基控制基础HIF-1αmRNA水平
(a)用所示的siRNA寡核苷酸转染HEK-293细胞,并进行qRT-PCR。直方图描绘了归一化为肌动蛋白mRNA水平的HIF-1αmRNA的相对水平。平均值+标准差至少从三个独立实验中计算得出。与对照相比,进行了Student's
t检验,* P <0.050和** P <0.010。实际的P值为:RelA,P = 0.012; RelB,P = 0.012; m / z。 c-Rel,P
= 0.076; p50,P = 0.348; p52,P = 0.035; HIF-1α,P = 0.007。
(b)使用所示抗体进行ChIP分析,并使用针对HIF-1α启动子和HIF-1α控制区的特异性引物进行PCR分析。
NF-κB调节常氧状态下的HIF-1α蛋白水平
缺氧后,HIF-1α主要在蛋白质水平受到调节[22]。由于本文的研究是在正常氧气水平下进行的,因此不易抑制PHD活性。为了研究NF-κB是否也可以调节HIF-1α蛋白水平,使用了NF-κB过表达和siRNA介导的敲低。鉴于HIF-1α蛋白在正常的氧气水平下会迅速降解,因此预期只有很小的变化。当所有NF-κB亚基都过表达时,有可能观察到蛋白质水平的增加(图3a)。但是,RelA和c-Rel诱导的增幅最高,其次是p50,p52和RelB。当使用蛋白酶体抑制剂MG132时,HIF-1α蛋白的变化最明显(图3a)。相反,当RelA,RelB和p52耗尽时,可以观察到HIF-1α蛋白水平的少量降低。当c-Rel耗尽时,也有很小的作用,但p50耗尽则未检测到作用(图3b)。无论是过表达还是siRNA介导的NF-κB亚基的敲除,HIF-1β的水平都保持稳定(图3)。
包含图片,插图等的外部文件。
对象名称是bic874i003.jpg
在单独的窗口中打开
图3
NF-κB亚基控制HIF-1α蛋白的基础水平
(a)用所示的DNA构建体转染HEK-293细胞,并在转染后48小时获得全细胞裂解物。收获前3小时,将一半样品与50μMMG132孵育。然后在这些提取物上进行指定蛋白质的蛋白质印迹分析。
(b)用所示的siRNA寡核苷酸转染HEK-293细胞,并在转染后48小时获得全细胞裂解物。收获前3小时,将一半样品与50μMMG132孵育。然后在这些提取物上进行指定蛋白质的蛋白质印迹分析。
TNFα诱导的NF-κB调节HIF-1α表达
NF-κB的激活与炎症反应最相关[13]。它会在细胞因子(例如TNFα)或细菌产物(例如LPS(脂多糖))之后迅速激活。实际上,尽管还没有完全阐明其机制,但已经描述了这些刺激均能激活HIF-1α[23,24]。鉴于许多炎症性疾病和某些癌症中存在长时间的TNFα暴露,我们研究了HIF-1α是否在这种条件下被诱导(图4)。我们将细胞暴露于TNFα几个小时,并分析了核部分(图4a)。可以检测到所有NF-κB亚基的核蓄积,并且还可以检测到HIF-1α的显着增加(图4a)。此外,这种延长的TNFα治疗还诱导了两个HIF靶基因GLUT1和3的增加(图4b)。为了测试观察到的HIF-1α,GLUT1和GLUT3蛋白的增加是否是由于较高的mRNA水平引起的,在TNFα处理后的指定时间内进行了qRT-PCR。有趣的是,有可能观察到HIF-1α,GLUT3和GLUT1
mRNA的增加,而HIF-2αmRNA却没有变化(图4c)。这些结果表明,HIF-1α和GLUT3蛋白的增加是由于基因转录的增加,而HIF-2α水平的增加可能是由于蛋白质稳定性的影响。此外,siRNA介导的HIF-1α或HIF-2α耗竭表明TNFα诱导的GLUT3是HIF-1α依赖性的(图4d和补充图S1,网址为http://www.BiochemJ.org/bj/412/bj4120477add)。
htm),表明TNFα诱导的HIF-1α具有转录活性。
包含图片,插图等的外部文件。
对象名称是bic874i004.jpg
在单独的窗口中打开
图4
TNFα治疗诱导NF-κB和HIF-1α
(a)用10 ng / mlTNFα处理HEK-293细胞指定的时间,制备核提取物,并通过Western blot分析这些提取物中的指定蛋白质。
PCNA被用作加载控件。 (b)按(a)中所述处理细胞,并通过Western印迹分析细胞提取物以确定的HIF-1α靶标。
(c)按(a)中所述处理细胞,但是提取mRNA并针对指示的基因转录进行qRT-PCR。直方图描绘了标准化为肌动蛋白mRNA水平的特定mRNA转录物的相对水平。平均值+标准差至少从三个独立实验中计算得出。
(d)在总mRNA提取之前24小时,用所示的siRNA寡核苷酸转染细胞并用10ng / mlTNFα处理。如(c)中那样进行qRT-PCR。
TNFα诱导的HIF-1α活性是NF-κB依赖性的
为了测试观察到的HIF-1α变化是否是NF-κB依赖性的,将不同NF-κB的siRNA与TNFα处理结合使用。除p105 /
p50外,所有亚基的消耗均导致TNFα诱导的HIF-1α表达受损(图5a)。此外,HIF-1α的消耗导致PHD2的TNFα诱导受损(补充图S2,位于http://www.BiochemJ.org/bj/412/bj4120477add.htm),表明这是HIF-1α依赖性事件。此外,还检测到HIF-1α诱导招募到其某些靶基因的启动子(图5b)。
包含图片,插图等的外部文件。
对象名称是bic874i005.jpg
图5
TNFα以NF-κB依赖性方式诱导HIF-1α活性
(a)HEK-293细胞用指定的siRNA寡核苷酸转染,在收获前24小时用10ng /
mlTNFα处理,并制备全细胞裂解物。使用指定的抗体通过蛋白质印迹分析提取物。
(b)使用指定的抗体进行ChIP分析,并对HIF-1α靶基因的特定区域VEGF,CA9和DEC1进行PCR。 HRE,缺氧反应元件。
(c)与(b)相同,但分析了HIF-1α启动子和对照区。 (d)按(a)中所述处理细胞,并使用所示抗体进行蛋白质印迹分析。 PCNA被用作加载控件。
为了验证TNFα诱导HIF-1α启动子的改变,进行了ChIPs。在TNFα治疗18小时后,有可能观察到RelA和p52主动募集到HIF-1α启动子(图5c)。重要的是,TNFα处理不会诱导NF-κB与HIF-1α基因的控制区结合。此外,RelA,RelB,c-Rel和p52的消耗减少了TNFα诱导的HIF-1α介导的PHD2诱导(图5d)。这些结果表明,TNFα诱导的HIF-1α水平和活性的变化是NF-κB依赖性的。
完全阻断NF-κB激活途径可防止缺氧诱导的HIF-1α
鉴于NF-κB可以控制基础HIF-1αmRNA水平,我们研究了阻断NF-κB是否可以预防缺氧诱导的HIF-1。我们利用基因敲除细胞的优势,并将siRNA用于调节NF-κB的上游激酶复合物。我们可以观察到,当通过基因敲除或siRNA介导的沉默使IKKα或IKKβ耗尽时,对HIF-1α水平的影响很小(图6a和6b),6b),这表明蛋白质稳定可以补偿规范或非规范途径的任何损害。但是,当IKKα和IKKβ均被耗尽时,对缺氧反应的HIF-1稳定性会严重受损(图6c)。这表明HIF-1α基因转录是由NF-κB的几个亚基介导的,这些亚基源自两种激活途径。这些结果支持了我们关于NF-κB如何控制HIF途径的mRNA和ChIP结果。鉴于缺氧不会诱导HIF-1αmRNA的增加,因此在缺氧处理后未观察到NF-κB募集到HIF-1α启动子的变化也就不足为奇了(图6d)。再次,我们无法检测到任何与HIF-1α基因控制区结合的NF-κB(图6d)。这些结果表明,在缺乏NF-κB的情况下,低氧后HIF-1α稳定所见的任何损伤是由于基础mRNA水平的变化,而不是由于缺少对mRNA的主动诱导。
包含图片,插图等的外部文件。
对象名称是bic874i006.jpg
在单独的窗口中打开
图6
完全抑制NF-κB会削弱缺氧诱导的HIF-1α水平
(a)将U2OS细胞用IKKα或IKKβsiRNA寡核苷酸转染,并在收获前在指定的时间内暴露于1%O2。通过Western印迹分析全细胞裂解物。
(b)在指定的时间内,将野生型,IKKα-/和IKKβ-/-小鼠胚胎成纤维细胞暴露于1%O2,制备全细胞裂解液。通过Western印迹分析提取物。
(c)在指定的时间内,将野生型和IKKα/β-/-小鼠胚胎成纤维细胞暴露于1%O2,制备全细胞裂解物。通过Western印迹分析提取物。
(d)在收获前,不对U2OS细胞进行处理或用1%O2处理4
h,然后使用所示抗体进行ChIP分析。使用针对HIF-1α启动子和HIF-1α控制区的特异性引物进行PCR分析。
讨论
慢性炎症是自我延续的,并且由于异常活跃的转录因子的作用,已证明其会扭曲微环境。细胞环境中生长因子,趋化因子,细胞因子和ROS平衡的随之变化,为癌症和转移从头发展所需的生长和存活轴提供了依据[25]。因此,我们假设在此过程中存在两个关键转录因子NF-κB和HIF之间的新型相互作用。在本研究中,我们证明了HIF-1α基础mRNA水平受几个NF-κB亚基控制(图1-3、55和6).6)。此外,我们显示了TNFα-(在一定程度上是低氧)诱导的NF-κB调节HIF-1α的水平和活性,从而触发诸如GLUT3和PHD2等靶基因的转录(图4-6)。
本研究的结果对许多类NF-κB和HIF-1失控的病理学具有许多意义,例如类风湿性关节炎或癌症。但是,尽管有这些暗示,但HIF-1α的NF-κB调节可能是刺激特异性的,甚至是细胞类型特异性的。证据来自可用的基因敲除模型。虽然HIF-1α缺失会在第9-11天导致胚胎致死率[26,27],且缺陷早在7天就已显现,但IKKα和IKKβ的基因遗传缺失会在第12天造成致死率[28]。
IKKα,IKKβ或RelA的单缺失会在更晚的阶段导致致死性[28]。在HIF-1α-/-小鼠中,在脑中膜中发现了神经管形成缺陷,心血管畸形和细胞死亡增加。有趣的是,IKKα/β双无效小鼠在神经功能上也有缺陷,但这些缺陷似乎与神经元上皮细胞凋亡增加有关。重新分析这些小鼠胚胎在不同组织中的HIF-1α表达将是很有趣的。
本研究的结果还表明,尽管存在氧气,TNFα仍可诱导HIF-1活性。 TNFα可以在细胞中产生ROS
[29],并且已经证明它们可以抑制PHD活性[30,31]。尽管我们尚未研究这种可能性,但有关HIF-1α稳定性和活性的当前结果将支持这一点。精确的ROS测量和PHD活性测定将回答这些问题。
Regulation of hypoxia-inducible factor-1α by NF-κB
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2474706/
https://openi.nlm.nih.gov/detailedresult?img=PMC1064874_ar1447-4&req=4
氧代谢在类风湿关节炎的发病机理中具有重要作用。细胞氧化磷酸化过程中产生的活性氧(ROS),以及在氧化爆发期间由吞噬细胞活化而产生的活性氧,超过了生理缓冲能力并导致氧化应激。
ROS的过量产生会损坏蛋白质,脂质,核酸和基质成分。它们还用作重要的细胞内信号分子,可放大滑膜的炎症反应。假定与滑膜灌注变化有关的缺氧和复氧的重复周期可激活缺氧诱导因子-1α和核因子-κB,这两个关键的转录因子受细胞氧合和细胞因子刺激的变化所调节,并进而协调对滑膜炎持续存在至关重要的一系列基因的表达。对这些途径中涉及的复杂相互作用的理解可能允许发展类风湿关节炎的新治疗策略。
底线:过量产生的ROS会破坏蛋白质,脂质,核酸和基质成分,它们还作为重要的细胞内信号分子,放大了滑膜的炎症增殖反应。低氧和复氧的重复循环与滑膜灌注的变化有关被认为激活缺氧诱导因子-1α和核因子-κB,这两个关键的转录因子受细胞氧合和细胞因子刺激的变化调节,进而协调了对滑膜炎持续性至关重要的一系列基因的表达。
Immunologic Consequences of Hypoxia during Critical Illness | Anesthesiology
| ASA Publications
https://anesthesiology.pubs.asahq.org/article.aspx?articleid=2524652
危重疾病缺氧的免疫学后果
Immunologic Consequences of Hypoxia during Critical Illness
Anesthesiology (H.D.K., G.-J.S., M.K.), and the Radboud Centre
for Infectious Diseases (H.D.K., J.G.v.d.H., P.P., M.K.), Radboud University
Medical Center, Nijmegen, The Netherlands; and Organ Protection Program,
Department of Anesthesiology, University of Colorado School of Medicine, Aurora,
Colorado (H.K.E.).
摘要
缺氧和免疫力在临床,细胞和分子水平上高度交织在一起。预防组织缺氧和调节全身性炎症是重症监护病房日常工作的基石。缺氧的免疫学作用可能会促进预后并可能代表治疗目标。缺氧和下游信号通路的激活导致先天免疫反应增强,旨在增加病原体清除率。另一方面,低氧还对淋巴细胞和其他组织发挥抗炎和组织保护作用。尽管关于缺氧的净免疫学作用和下游通路的药理学调节的人类数据有限,但临床前数据支持通过危重患者中氧状态的调节或缺氧信号通路的药理学调节来定制免疫应答的概念。

摘要
缺氧会影响分子和细胞的炎症过程,从而可能影响严重疾病的预后。在缺氧单一途径中调节氧合作用和药物干预可以支持这些患者的免疫功能。
优化氧合以防止组织缺氧是重症监护的基石之一。同时,在重症监护病房(ICU)住院的大多数患者中,发生炎症过程,这可能会影响预后。由于缺氧和免疫在分子,细胞和临床水平上高度相关,因此缺氧的免疫学作用可能代表危重患者的治疗目标。在细胞水平上,缺氧激活不同的缺氧信号通路,包括一组称为缺氧诱导因子和腺苷信号传导的转录因子。体外和动物研究表明,这些途径与炎症反应的调节有关,动物研究表明,这些途径与重症患者经常遇到的炎症(如败血症1,2和肺损伤)相关。3
,4此外,炎症性疾病的特征通常在于组织缺氧,这是由于代谢需求增加以及水肿,微血栓和肺不张导致的代谢底物减少,进而引起“炎症性缺氧”。5,6因此,针对特定的在重症患者的一系列炎症条件下,组织氧合水平可能是有利的。或者,这些作用也可以通过针对缺氧信号通路的药物干预来实现。
在当前的审查中,我们概述了缺氧的免疫学后果。我们专注于有关重症患者炎症状况的体外,动物和人体研究,包括对氧依赖性信号传导途径和中间信号传导系统(例如,低氧诱导因子[HIF]系统和腺苷代谢)的讨论。
2,7此外,我们讨论了干预这些机制的临床潜力,包括有关高氧症潜在弊端的证据,治疗性允许性低氧的可行性以及作用于氧依赖性途径的药物疗法。低氧和HIF在重症患者炎症状况范围之外的作用在其他地方进行了综述。2,7–9
缺氧的免疫学作用
缺氧的免疫学作用证据主要是在使用髓样细胞的体外研究中建立的(表1)。10-12长期缺氧已被证明本身就是一种炎症刺激,长期的缺氧会导致组织中细胞因子的产生。人类巨噬细胞系。14此外,在原代人单核细胞中,低氧增加毒素脂多糖或植物血凝素的刺激后,会增加促炎细胞因子的产生。13,15相反,其他研究表明,低氧会使促炎特性(M1-like
18,19除了这些矛盾的发现外,这些体外研究也难以解释,因为控制条件通常是室内空气,与生理组织Pao2相比,Pao2更高。尽管如此,这些体外研究表明,氧合作用具有免疫学作用,尽管这种反应的方向可能取决于细胞类型和激活状态。
表格1。
低氧对免疫的影响的体外和临床前研究
低氧对免疫的影响的体外和临床前研究
全尺寸|幻灯片(.ppt)
健康的体内缺氧志愿者表现出增强的离体中性粒细胞趋化性,吞噬作用和活性氧的产生24,并增强了活化B细胞的κ-轻链增强子(NF-κB)关键炎症转录因子核因子的活性。
25此外,健康受试者高海拔低氧(动脉血氧饱和度[Sao2],75至90%)暴露4天会导致促炎性白细胞介素-6、21、22的血浆水平升高,同时缺氧时间更短不要诱发这种全身性反应17,23(表1)。总而言之,体内长时间缺氧会增加骨髓细胞的炎症反应,并引发全身性免疫反应。
关于潜在的机制,体外缺氧诱导了广泛的细胞过程级联反应,其受氧敏感性途径(包括脯氨酰羟化酶(PHD),转录因子HIF和NF-κB,腺苷信号传导途径以及其他对氧敏感的过程)调控。这些细胞机制提供了对有限氧气供应条件的适应性,并且每种途径以不同的方式促进了缺氧的免疫学作用。这可以解释为什么缺氧会同时引起促炎和抗炎以及组织保护作用,如下文进一步详述。
HIF-1α的调控
HIFs代表一组转录因子,可响应缺氧而介导大量细胞适应。26HIFs是由HIF-β和三个氧依赖性转录活性α亚基之一(HIF-1α,HIF-2α和HIF)组成的异二聚体。
HIF-3α,其中HIF-1α是研究最广泛的同工型。在图1中详细说明了在常氧,低氧和炎症条件下负责调节HIF-1α蛋白稳定和信号传导的细胞机制。在常氧条件下,氧依赖性PHD-1,PHD2和PHD3以及天冬酰胺基羟化酶抑制因子的HIF(FIH)羟基化HIF-1α,之后羟基化的HIF-1α与Von
Hippel-Lindau复合物结合。 HIF与Von
Hippel-Lindau的结合最终导致蛋白酶体的泛素化和降解。在缺氧条件下,氧依赖性羟化酶是无活性的,从而防止了HIF-1α的降解。因此,缺氧以翻译后的方式调节HIF-1α。
HIF-1α调节的第二个独立于氧的翻译后机制涉及热休克蛋白(HSP)90。HSP是细胞应激反应的关键参与者,起分子伴侣蛋白的作用,可促进其构象,定位和多种功能蛋白质。
HSP90阻止了HIF-1α的不依赖氧的降解,从而导致HIF-1α的稳定化。27-29此外,HSP90与HIF-1α的结合有助于与HIFβ偶联并随后进行反式激活。29
图。1。
常氧,低氧和炎症条件下的缺氧诱导因子(HIF)-1α调节和信号转导。
HIF-1α亚基不断产生,但在常氧条件下迅速降解。已经描述了HIF-1α调节的几种途径。首先,在常氧条件下,HIF-1α亚基被氧依赖性脯氨酰羟化酶结构域酶(PHD)迅速羟基化,随后被泛素连接酶Von
Hippel-Lindau(VHL)蛋白捕获并被蛋白酶体降解。其次,氧依赖性天冬酰胺基羟化酶因子抑制HIF(FIH)将保守的天冬酰胺基残基羟基化,阻止了共激活因子P300和cAMP反应元件结合蛋白(CBP)的募集,进而抑制了HIFβ的二聚作用。在缺氧期间,PHD和FIH活性降低,导致HIF-1α亚基在细胞质中积聚。活化的C激酶1(RACK1)和热休克蛋白90(HSP90)的受体以不依赖氧的方式调节HIF-1α:RACK促进不依赖氧的HIF-1α的蛋白酶体降解,而HSP90与RACK竞争,从而稳定HIF
-1α,并促进其反式激活。积累后,HIF-1α被P300 /
CBP共激活并与HIFβ二聚形成稳定的HIF-1αβ二聚体。这些二聚体易位至细胞核并与基因启动子增强子区域的缺氧反应元件(HRE)结合,从而导致转录活性。
HIF-1α稳定导致许多(大于100个)缺氧反应性基因转录。由于FIH在低于PHD的氧气浓度下仍保持活性,因此FIH抑制了HIF-1α蛋白的活性,该蛋白在中等氧不足时可以避免破坏。不仅缺氧,而且暴露于细菌和细菌产品(如脂多糖(LPS))也会导致HIF-1α积累。
NF-κB=活化的B细胞的κ轻链增强子的核因子; TLR =收费型受体。
最后,HIF-1α的转录和翻译通过炎症刺激而增加。因此,缺氧,细胞应激和炎症(协同)增强了HIF-1α的稳定性。2,7
HIF-1α的稳定化促进了100多个缺氧反应性基因的转录,其中30个导致缺氧适应,例如促红细胞生成素和血管内皮生长因子。31尽管HIF-1α的自动调节系统尚未完全阐明,但仍存在32在体外,低氧以剂量依赖的方式诱导HIF-1α表达,但是长时间的低氧会导致靶向HIF(aHIF)的微小RNA介导的HIF-1α下调。
33,与体外数据相反,在体外数据中,仅显示低氧可预防HIF-1α降解,而体内低氧刺激HIF-1α转录,随后降低至基线水平,
34人类研究表明,由于缺氧,白细胞HIF-1α表达35和下游靶基因表达36的个体间差异很大,这可能是由于aHIF介导的负反馈所致。研究HIF调节的表型差异。
HIF-1α和腺苷信号参与缺氧免疫作用
缺氧,HIF和NF-κB之间的分子相互作用
HIF-1α和NF-κB(后者被认为是炎症反应的主要调节剂)之间的调节高度交织。37,38在失活状态下,NF-κB与细胞质中的抑制蛋白IκBα结合。不仅炎症刺激而且其他信号也会激活酶IκB激酶(IKK),导致IκBα磷酸化。随后,NF-κB移位进入细胞核,并产生以炎症细胞因子产生为特征的炎症反应。39NF-κB活性的另一个下游作用是增强HIF-1α的转录。40-42相反,HIF-1α活性增强了NF通过增加IKK和NF-κB亚基p65.41,43的丰度来激活-κB活性。此外,缺氧可阻止PHD依赖的IKK降解。44体外研究证实了这种作用,因为联合抑制PHD-1和FIH可增强基础NF-矛盾的是,PHD-1和FIH抑制在炎性条件下抑制NF-κB活性。45这些数据说明缺氧,氧依赖性羟化酶,HIF-1α,和NF-κB。此外,如前所述,效果取决于细胞活化状态。
HIF-1α的细胞和体内免疫作用
在细胞水平上,免疫细胞中HIF-1α的稳定导致分化反应,这在很大程度上取决于细胞类型。在嗜中性粒细胞中,参与上皮嗜中性粒细胞结合的β2-整合素的诱导,16病原体结合嗜中性粒细胞胞外陷阱的调控以及抗菌活性46都是HIF-1α依赖性的。46HIF-1α稳定抑制巨噬细胞和嗜中性粒细胞的凋亡43,47,并且48HIF-1α还会导致toll样受体449的表达增加,并增强巨噬细胞的吞噬作用50和细菌杀灭作用51。
使用细胞特异性转基因敲除小鼠和药理性HIF-1α调节的大量动物研究也证明了HIF-1α对细胞类型的作用。髓样HIF-1α敲除小鼠在链球菌皮肤感染中的发病率高于野生型同窝仔鼠,这表明髓样细胞中的HIF-1α对于启动清除局部感染所需的炎症反应是必不可少的。51脂多糖(用于模拟革兰氏阴性感染)52或脂蛋白酸和肽聚糖(用于模拟革兰氏阳性感染),53缺乏髓样HIF-1α的小鼠的炎症反应减弱,与组织损伤减少和存活率提高相关。52
,HIF-1α的功能获得导致无菌性和细菌性腹膜炎中的压倒性炎症反应,加重了器官损伤并损害了存活率。1因此,在髓样细胞中,HIF-1α对于产生有效的炎症反应至关重要。感染的同时,HIF-1α的过度表达导致败血症早期,炎性阶段的临床表现老鼠。
与在髓样细胞中观察到的促炎作用相反,HIF-1α活性在淋巴细胞中诱导抗炎和组织保护作用。例如,HIF-1α诱导导致调节性T细胞数量增加,并由于炎症减弱而提供后续的组织保护。54此外,在鼠细菌性腹膜炎模型中,T细胞特异性HIF-1α缺乏症导致HIF-1α缺乏水平升高。
55提示HIF-1α对B细胞具有抗炎作用,在给予小鼠脂多糖之前,用二甲基草酰甘氨酸抑制PHD可导致B1细胞产生白介素10增强,从而使巨噬细胞偏向抗炎M2样表型。56此外,其他研究表明,通过诱导HIF靶基因FoxP3.54来诱导抗炎调节性T细胞分化的转录程序处于HIF的控制之下
除了对专用免疫细胞的作用外,HIF-1α稳定作用还对其他细胞(例如肠和肺泡上皮和心肌细胞)产生免疫作用。通过抑制PHD抑制鼠化学性结肠炎中HIF-1α的药理作用可降低TNFα,白细胞介素6和白细胞介素1β的水平,而抗炎性白细胞介素10的水平则提高57,临床结果也有所改善。58,59类似地,药理学呼吸机诱发的肺损伤中的PHD抑制导致HIF-1α依赖性的减轻肺损伤并延长生存期,而HIF-1α抑制则加重肺损伤并缩短生存期。4HIF-1α的组织保护作用还参与了针对缺血性损伤。例如,通过远程缺血预处理对心肌的保护取决于通过HIF-1α,60、61介导的白介素10产量的增加,并且心肌HIF-1α的表达介导了糖酵解的代谢转换,这对于适应缺血至关重要。62表2.45,63–74提供了PHD抑制作用的体外和动物研究的免疫学作用
表2。
PHD抑制剂对免疫的影响的体外和临床前研究
PHD抑制剂对免疫的影响的体外和临床前研究
全尺寸|幻灯片(.ppt)
总的来说,髓样细胞中的HIF-1α活性参与了旨在清除病原体的免疫反应,而淋巴细胞,上皮细胞和肌细胞中的HIF-1α活性诱导了抗炎和组织保护作用(概述见图2)。
。尽管这些相反的作用看似矛盾,但在肿瘤学领域的研究表明,髓样HIF-1α活性抑制T细胞反应。75因此,可以想象,在炎症和感染的情况下,不同免疫细胞之间的局部相互作用需要优化感染控制并同时防止组织损伤76。
图2。
缺氧与炎症之间的相互作用。缺氧可增强免疫反应,本身就是一种炎症刺激。缺氧导致缺氧诱导因子1α(HIF-1α)的细胞稳定,从而与活化B细胞的κ轻链增强子(NF-κB)的关键炎症转录因子核因子产生协同作用。另外,炎症增强了HIF-1α的转录和翻译,从而在缺氧和炎症的情况下产生了协同作用。在中性粒细胞和单核细胞等髓样细胞中,HIF-1α活性具有促炎作用,旨在清除病原体。相反,在许多其他细胞(例如T细胞,肺和间质上皮以及肌细胞)中,HIF-1α活性具有抗炎作用。此外,低氧通过腺苷途径发挥抗炎作用,因为它增加了腺苷祖细胞三磷酸腺苷(ATP)和二磷酸腺苷(ADP)的利用率,上调转化酶CD39和CD73以增强腺苷生成,并增加抗炎药的表达腺苷2A和2B受体(A2A和A2B)。
CD73和腺苷受体的上调是HIF-1α依赖性的。 IL-10 =白介素10; TLR4 =收费型受体4。

败血症中的HIF-1α
HIF-1α在脓毒症中的作用尤其令人关注,因为炎症和组织缺氧经常并存,后者是由于氧气需求和可用性不匹配所致。败血症早期的免疫宿主反应的特征是(过量)促炎细胞因子的产生,其目的是清除病原体,但也导致败血性休克的临床综合征。然而,同时进行抗炎反应,大概是为了减少促炎反应,从而防止附带组织损伤。当过于明显和/或持续时,这种抗炎反应会导致免疫系统的状态被严重抑制。人们日益认识到,这种现象被称为“败血症诱导的免疫麻痹”,使患者更容易遭受继发感染,并且是败血病患者晚期死亡的主要因素。77
根据先前描述的数据,HIF-1α活性可能增强促炎作用和先天免疫功能,这可能对败血症诱发的免疫麻痹有益。小鼠慢性慢性轻度缺氧可部分逆转与脓毒症诱导的免疫麻痹相似的内毒素耐受性,这一观点得到了支持。20但是,这项单动物研究并未完全反映人体内HIF-1α的复杂动态。败血症。此外,需要强调的是,已经在体外和动物中进行了上述关于炎症和缺氧之间的相互作用的研究。从动物研究到人类处境的翻译是一个重要的辩论主题。78,79
幸运的是,最近在败血症患者中进行的两项观察性研究增加了我们对败血症期间HIF-1α动态的了解。其中一种是在入院后2至4
h内获得样本,单核细胞中HIF-1αmRNA表达增加。80此外,HIF-1α诱导了Toll样受体调节剂白介素1受体相关的负性激酶M,
80与此相反,另一项研究发现白细胞HIF-1α蛋白和mRNA表达降低,但在较晚的时间点(即入院后24小时之内)获得了样本。81尽管在解释数据时必须谨慎从临床患者研究的临床前工作中可以预见,早期的促炎反应会驱动HIF-1α表达增加,从而导致负调节剂(如白介素1受体相关激酶M)的诱导,以抵消过度的炎症,最终导致败血症后期HIF-1α水平降低。
通过腺苷途径的组织保护和抗炎作用
缺氧还可以通过腺苷途径发挥抗炎和组织保护作用,据报道其中一些元素是HIF-1α依赖性的。82,83
细胞窘迫(例如,hypoxia84)会导致腺苷祖细胞三磷酸腺苷和二磷酸腺苷的可用性增加。85低氧导致CD39(胞苷磷酸酶)上调,86,87将三磷酸腺苷和二磷酸腺苷转化为单磷酸腺苷,
HIF-1α依赖性上调CD73(5'-外核苷酸酶),将单磷酸腺苷转化为腺苷。82这些酶的组织保护作用已在使用敲除小鼠的研究中得到证实。例如,缺乏CD39或CD73的小鼠在炎性或缺血性损伤后显示出更高的发病率和死亡率。88-90相应地,HIF-1α的基因过表达导致CD73的上皮表达增加,并改善了鼠化学性结肠炎的结局。91最后,缺氧会增加HIF-1α依赖性腺苷2A(A2A)和2B(A2B)受体的表达。83刺激这些受体会在鼠缺血再灌注模型中产生全身性抗炎作用89缺氧,
92和炎症[93]。此外,在急性肺损伤的小鼠模型中,允许的低氧(吸入氧气的分数[Fio2],10%)可以减轻肺损伤并提高生存率,94并诱导A2B-呼吸机诱发的肺损伤期间1型肺泡细胞中的受体被证明依赖于HIF-1α3。同样,低氧预适应以A2B受体依赖性方式保护小鼠免于肝缺血和再灌注损伤。95
有限的可用人类数据证实了缺氧导致腺苷可用性提高。例如,在健康志愿者中暴露于短期缺氧(20分钟,Sao2,80%)会增加血浆腺苷水平96。此外,一些人体实验研究表明,静脉内给予腺苷97以及口服治疗可改善腺苷信号传导的抗炎作用。腺苷摄取抑制剂双嘧达莫98减弱了实验性人类内毒素血症期间促炎性白细胞介素6的反应,双嘧达莫治疗也增加了抗炎性白细胞介素10的产生。98但是,在这些后者的研究中,腺苷的可利用性不是由低氧引起的。最后,一项概念验证的临床研究表明,干扰素-β-1a增强人肺组织中的CD73表达,并且向急性呼吸窘迫综合征(ARDS)患者施用这种细胞因子与白细胞介素6和白细胞介素8减少有关。浓度以及改善的Pao2
/ Fio2比率和存活率99。
除了HIF-1α,NF-κB和腺苷代谢和信号传导途径外,还确定了其他对氧敏感的转录因子,尽管尚未完全阐明确切的氧依赖性机制和下游效应(参见参考文献100)。
缺氧与炎症之间复杂相互作用的示意图如图2所示。
重症患者缺氧
缺氧性呼吸衰竭是ICU患者的常见病状,根据定义(通常需要机械通气和/或Pao2 / Fio2比值小于300
mmHg101-103),其发生率为22%至33%101,102,并且死亡率为31%至52%。101-103低氧性呼吸衰竭的亚类为ARDS,占呼吸衰竭患者的3%至70%。101-103ARDS的严重程度可以根据柏林的定义归类为轻度(
200至300 mmHg),中度(100至200 mmHg)或重度(小于100
mmHg),死亡率介于32%至65%之间。103-109重要的是区分低氧性呼吸衰竭的诊断(即,缺氧引起的气管插管和机械通气的指示)和实际的缺氧(即低Pao2),因为低氧性呼吸衰竭患者的Pao2水平可以正常。重症监护病房入院时发生缺氧(即Pao2低于80
mmHg)的频率很高(40%)110,在荷兰重症监护病房患者中进行的一项回顾性队列研究显示,重症监护病房入院时或在重症监护病房期间缺氧与病情增加有关110即使在对疾病严重程度和其他混杂因素进行校正后,死亡率也是如此。110在澳大利亚和新西兰ICU患者中进行的类似分析也证实了ICU入院时低氧与死亡率增加之间的关联。111然而,这些研究本质上是观察性的,尽管努力在消除偏见方面,混杂因素可能仍然起作用。因此,这些研究不能用于指导氧疗。当前,缺乏用于重症患者的氧合目标。自从对ARDS潮气量进行具有里程碑意义的研究以来,ARDS研究使用的目标为55至80
mmHg或Sao2 88%至95%112,尽管没有确凿的证据支持这些目标113。
在ICU患者中进行氧合作用的临床试验结果对于确定患者不同亚组的最佳氧合作用目标是必要的。结果,O2-ICU研究将植入系统性炎症的ICU患者随机分配到目标Pao2为120或75毫米汞柱(Clinicaltrials.gov标识符NCT02321072)。和Fio2 100%,持续24个小时,并用等渗漏液和高渗悬浮液恢复,由于高氧/高渗组死亡率的临界值显着增加,因此被初步终止。114时间,ST段抬高型心肌梗死中高空气对氧试验显示,补充氧气治疗的常氧性ST抬高型心肌梗死患者表现出与没有额外的吸氧的常氧患者比例,肌酸激酶水平和心肌梗死面积增加了。115高氧在机械通气ICU患者中进行的一系列前后研究分别将Sao2指标为90%至92%116和92%至95%117,这与不良结局无关。另一随机对照试验研究最近证实了保守性氧合作用策略的安全性和可行性,将自由氧合策略(SpO2大于96%)与保守性氧合作用策略(SpO2为88%至92%)进行比较118。探索个性化氧气靶标以影响重症患者呼吸的方法。
重症患者低氧和炎症的临床前数据的翻译
正如动物研究和有限的临床数据所表明的那样,缺氧和下游信号通路可能代表重症患者炎性疾病如败血症和肺损伤的病理生理学中重要且可修正的因素。但是,在将这些见解转化为临床实践之前,仍然需要采取许多措施。重症患者在炎症条件下的宿主反应是复杂的,个体间存在相当大的差异,并且会随时间而变化。然而,可以设想的是,针对有针对性的,个性化的,有利的免疫表型调节免疫应答,例如败血症诱发的免疫麻痹中的免疫刺激治疗或急性肺损伤中的抗炎治疗,可能具有临床益处。119许多具体的治疗靶标干预措施未能在临床试验中显示出益处120,天真地认为靶向低氧依赖的途径是“神奇的子弹”。然而,优化所有可修改参数以使炎症宿主反应适应更优选的情况仍然是天真的。考虑过的。由于在ICU中每天都要进行氧气管理,因此也应考虑充氧的免疫作用,作为优化宿主反应的一种手段。
尽管完全基于体外和动物数据,但氧合依赖性免疫调节策略可以设想为通过预防缺氧来消除缺氧诱导的免疫学作用,或者通过预防高氧症甚至允许或诱发缺氧来增强缺氧的免疫学作用。例如,动物数据表明,避免高氧血症甚至允许缺氧在急性肺损伤中是有益的94,除了预防直接的氧中毒。自然,只有在考虑到安全裕度的情况下,才有意或无意将缺氧作为一种治疗策略,特别是当Pao2靶标位于氧-血红蛋白解离曲线的陡峭部分时。如先前提出的那样,合适的氧合作用监测和控制系统应使用有关脉搏血氧饱和度,组织氧合作用和动脉血氧张力的实时数据来实现预定义的氧合作用121,并且自然应该对其安全性,可行性和功效进行广泛的测试。
但是,应谨慎行事,因为长期神经认知功能障碍与ARDS患者缺氧的时间长短(即Sao2,小于90%)之间存在关联。122因此,低氧的短期益处,即需要仔细权衡炎症的假定治疗作用和长期作用,即神经认知损害。
除了缺氧,或者如果不能证明允许的缺氧,还可以通过药理抑制PHD来追求HIF-1α介导的作用。
PHD抑制剂FG-4497增强了小鼠的HIF-1α稳定性,随后具有干细胞对放射的抗性,123改善了肾脏移植的存活率,124以及减弱了结肠炎期间TNFα的表达和体重减轻。58一种类似的PHD抑制剂(FG-2216)导致血液透析患者血浆促红细胞生成素水平升高125;但是,由于致命肝坏死和其他患者出现异常肝酶测试的情况,美国食品药品监督管理局暂停了该临床试验,并中止了进一步的开发。126尽管如此,针对PHDs治疗贫血的新药的临床试验目前正在对患有慢性肾脏疾病和透析的患者进行治疗。26尚没有确定药理学的HIF刺激是否会影响人类的免疫反应。另外,常用的PHD抑制剂二甲基草酰甘氨酸和上述FG化合物是泛羟化酶抑制剂,并且对HIF-1α稳定化不是特异性的,这可能导致不良作用。例如,二甲基草酰甘氨酸还可以稳定HIF-2α,从而导致促红细胞生成素水平升高58,59,从而可能导致红细胞增多症。可以通过更特异性的PHD抑制剂(例如选择性PHD-1抑制剂AKB-4924)和/或局部给药而不是全身给药来避免这种情况69。
两者合计,尽管通过氧合或缺氧信号传导途径的药理学调节来调节免疫应答的概念很诱人,但仍然存在问题,即这种方法是否可行并会为患者带来临床益处。因此,非常有必要进行评估这些作用的假定治疗潜力的研究。此外,在ICU中炎症条件下的免疫调节疗法仍然面临许多挑战。例如,脓毒症的抗炎策略在过去并不成功[120],可能是因为它们使患者对继发感染的抵抗力增强,尽管这也可能是由于该患者人群的异质性很高。目前正在研究针对脓毒症的预防和/或逆转免疫麻痹的免疫刺激疗法。77与此同时,寻找可识别ICU患者当前“免疫状态”的标记的研究仍在进行中,可能会更好地鉴定可从免疫调节疗法中受益的患者。 127
结论
缺氧与免疫系统之间存在广泛的相互作用。缺氧和炎症协同诱导HIF-1α稳定化,从而导致针对感兴趣的病原体清除率提高的细胞作用,同时发生了抗炎和组织保护机制,例如通过增强的腺苷代谢和信号传导。这些效应的净效应高度依赖于细胞类型和激活状态。对这些缺氧驱动机制的见解促进了个性化氧合靶标的概念,以适应发炎危重患者的免疫反应。
然而,由于目前缺乏这些数据,因此开发这种策略需要探索低氧对人体内免疫应答的推定作用。此外,还需要对药理性HIF-1α稳定剂和作用于腺苷途径的药物进行进一步研究。在任何情况下,危重病人的最佳Pao2和氧气输送都可能取决于诊断和合并症,临床医生应意识到,他们的氧气疗法不仅会影响饱和度,还会影响炎症宿主反应。由于目前尚无关于最佳氧合的临床指南,因此,探索自由氧合与限制性氧合的可行性的正在进行的临床试验非常有必要,并且正在进行中。这些可以进一步为个体化氧合治疗铺平道路。
Immunologic Consequences of Hypoxia during Critical Illness
Abstract
Hypoxia and immunity are highly intertwined at clinical, cellular, and molecular
levels. The prevention of tissue hypoxia and modulation of systemic inflammation
are cornerstones of daily practice in the intensive care unit. Potentially,
immunologic effects of hypoxia may contribute to outcome and represent possible
therapeutic targets. Hypoxia and activation of downstream signaling pathways
result in enhanced innate immune responses, aimed to augment pathogen clearance.
On the other hand, hypoxia also exerts antiinflammatory and tissue-protective
effects in lymphocytes and other tissues. Although human data on the net
immunologic effects of hypoxia and pharmacologic modulation of downstream
pathways are limited, preclinical data support the concept of tailoring the
immune response through modulation of the oxygen status or pharmacologic
modulation of hypoxia-signaling pathways in critically ill patients.
Regulation of hypoxia-inducible factor-1α by NF-κB
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2474706
Jun 15, 2008 · Endogenous p65, RelB and p52 have the highest effect on HIF-1α
mRNA levels (Figure 2 a). Despite inducing a slight reduction in HIF-1α mRNA,
c-Rel depletion did not induce statistically significant effects (Figure 2 a).
To assess whether NF-κB is directly regulating HIF-1α mRNA levels, ChIP analysis
was performed.
Cited by: 547
Publish Year: 2008
Author: Patrick van Uden, Niall S. Kenneth, Sonia Roch
HIF-1α restricts NF-κB-dependent gene expression to ...
https://dmm.biologists.org/content/8/2/169
Feb 01, 2015 · Hypoxia and inflammation are intimately linked. It is known that
nuclear factor κB (NF-κB) regulates the hypoxia-inducible factor (HIF) system,
but little is known about how HIF regulates NF-κB. Here, we show that HIF-1α
represses NF-κB-dependent gene expression. HIF-1α depletion results in increased
NF-κB transcriptional activity both in mammalian cells and in the model organism
...
Cited by: 28
Publish Year: 2015
Author: Daniel Bandarra, John Biddlestone, Sharon Mu
TNFα induces HIF-1α expression through activation of IKKβ ...
https://www.researchgate.net/publication/26823574_TNFa_induces_HIF-1a_expression... Treatment of cells with the IKKbeta inhibitor Bay 11-7082 reduced the TNFalpha-induced HIF-1alpha expression, suggesting that IKKbeta is required in this signaling pathway.
The TNF-α/ROS/HIF-1-induced upregulation of FoxMI ... https://www.ncbi.nlm.nih.gov/pubmed/22831955 The TNF-α/ROS/HIF-1-induced upregulation of FoxMI expression promotes HCC proliferation and resistance to apoptosis. The proliferation-specific transcription factor Forkhead box M1 (FoxM1) acts as a master regulator of cancer cell growth and survival and plays an important role in the development of hepatocellular carcinoma. Cited by: 87 Publish Year: 2012 Author: Limin Xia, Ping Mo, Wenjie Huang, Lin Zhang, Yin
TNFα通过激活IKKβ诱导HIF-1α表达
TNFα induces HIF-1α expression through activation of IKKβ
抽象
转录因子缺氧诱导因子1alpha(HIF-1alpha)受氧的利用以及各种炎症介质(包括肿瘤坏死因子α(TNFalpha))的调节。早期工作表明,磷脂酰肌醇-3-激酶(PI3K)和有丝分裂原激活的蛋白激酶(MAPK)信号通路参与常氧条件下TNFalpha介导的HIF-1alpha的积累和激活。在这里,我们提供的证据表明IkappaB激酶beta(IKKbeta)是TNFalpha调节HIF-1alpha所必需的。我们发现,TNFα增强了在常氧或低氧模拟条件下各种乳腺癌细胞系中HIF-1alpha蛋白的表达,但对HIF-1alpha
mRNA水平的影响很小。在IKKbeta稳定的克隆和瞬时转染子中发现增加的HIF-1alpha表达,并且IKKbeta的消耗持续减少HIF-1alpha蛋白的量。用IKKbeta抑制剂Bay
11-7082处理细胞会降低TNFalpha诱导的HIF-1alpha表达,表明在此信号传导途径中需要IKKbeta。在IKKbeta基因敲除小鼠胚胎成纤维细胞中显示出血管内皮生长因子(VEGF)(HIF-1α的直接靶标)表达降低。
我们进一步证明了原发性人类乳腺癌样本中IKKbeta和VEGF表达之间呈正相关。
我们的发现表明TNFalpha诱导的HIF-1alpha积累是IKKbeta依赖性的,并且可能能够通过炎症信号进一步了解HIF-1alpha的调控。
TNFα induces HIF-1α expression through activation of IKKβ
Anderson Cancer Center, Texus University
Abstract
The transcription factor hypoxia-inducible factor 1alpha (HIF-1alpha) is
regulated by oxygen availability as well as various inflammatory mediators,
including tumor necrosis factor alpha (TNFalpha). Early work suggested that the
phosphatidylinositol-3-kinase (PI3K) and mitogen-activated protein kinase (MAPK)
signaling pathways are involved in TNFalpha-mediated HIF-1alpha accumulation and
activation under normoxic conditions. Here, we provide evidence showing that
IkappaB kinase beta (IKKbeta) is required for HIF-1alpha regulation by TNFalpha.
We found that TNFalpha enhances HIF-1alpha protein expression in various breast
cancer cell lines under either normoxic or hypoxia-mimicking conditions, but has
little effect on the HIF-1alpha mRNA level. Increased HIF-1alpha expression was
found in IKKbeta stable clones and transient transfectants, and depletion of
IKKbeta consistently reduced the amount of HIF-1alpha protein. Treatment of
cells with the IKKbeta inhibitor Bay 11-7082 reduced the TNFalpha-induced
HIF-1alpha expression, suggesting that IKKbeta is required in this signaling
pathway. Decreased expression of vascular endothelial growth factor (VEGF), a
direct target of HIF-1alpha, was shown in IKKbeta-knockout mouse embryonic
fibroblast cells. We further demonstrated a positive correlation between IKKbeta
and VEGF expression in primary human breast cancer specimens. Our findings
indicate that TNFalpha-induced HIF-1alpha accumulation is IKKbeta dependent, and
may enable further understanding of the HIF-1alpha regulation by inflammatory
signals.
TNFα induces HIF-1α expression through activation of IKKβ | Request PDF
https://www.researchgate.net/publication/26823574_TNFa_induces_HIF-1a_expression_through_activation_of_IKKb
TNF-alpha诱导的NF-κB激活通过激活HIF-1 alpha刺激骨骼肌糖酵解代谢
TNF-alpha-Induced NF-kappa B Activation Stimulates Skeletal
Muscle Glycolytic Metabolism Through Activation of HIF-1 alpha
Maastricht University
摘要
在慢性阻塞性肺疾病(COPD)中,四头肌肌肉代谢特征向氧化代谢减少和糖酵解增加的转变是一致的发现。已经提出了慢性炎症作为这种病理性代谢适应的触发。实际上,促炎性细胞因子肿瘤坏死因子α(TNF-alpha)通过激活核因子κB(NF-kappaB)途径损害肌肉氧化代谢。
但是尚不清楚对肌肉糖酵解的推定作用。我们假设TNF-alpha诱导的NF-κB信号通过糖酵解调节因子缺氧诱导因子1alpha(HIF-1alpha)的激活来刺激肌肉糖酵解代谢。用TNF-α刺激野生型C2C12和C2C12-IkappaBalpha-SR(阻断的NF-kappaB信号传导)肌管,并研究其对糖酵解代谢的影响和本文HIF途径的参与。作为原理的证明,在先前特征明确的严重COPD和健康匹配对照组的临床稳定患者队列中,研究了股四头肌肌肉活检中HIF信号成分的表达。
TNF-α以依赖于NF-κB的方式增加了肌管葡萄糖的摄取和乳酸的产生,并增强了肌肉糖酵解代谢的多种效应子的活性和表达水平。此外,TNF-α激活了HIF信号传导,这需要经典的NF-kappaB激活。而且,HIF-1α的敲低大大减弱了TNF-α诱导的糖酵解代谢增加。因此,与对照相比,COPD患者的肌肉活检中HIF-1α和HIF-1α靶基因血管内皮生长因子(VEGF)的mRNA水平升高,这在肌肉TNF-α水平高的患者中最为明显。
TNF-α诱导的经典NF-kappaB激活以HIF-1alpha依赖性方式增强肌肉糖酵解代谢。
TNF-alpha-Induced NF-kappa B Activation Stimulates Skeletal Muscle Glycolytic
Metabolism Through Activation of HIF-1 alpha
Abstract
A shift in quadriceps muscle metabolic profile towards decreased oxidative
metabolism and increased glycolysis is a consistent finding in chronic
obstructive pulmonary disease (COPD). Chronic inflammation has been proposed as
a trigger of this pathological metabolic adaptation. Indeed, the
pro-inflammatory cytokine tumor necrosis factor alpha (TNF-alpha) impairs muscle
oxidative metabolism through activation of the nuclear factor kappa B
(NF-kappaB) pathway. Putative effects on muscle glycolysis however are unclear.
We hypothesized that TNF-alpha-induced NF-kappaB signaling stimulates muscle
glycolytic metabolism through activation of the glycolytic regulator
hypoxia-inducible factor-1alpha (HIF-1alpha). Wild-type C2C12 and
C2C12-IkappaBalpha-SR (blocked NF-kappaB signaling) myotubes were stimulated
with TNF-alpha and its effects on glycolytic metabolism and involvement of the
HIF pathway herein were investigated. As proof-of-principle, expression of HIF
signaling constituents was investigated in quadriceps muscle biopsies of a
previously well-characterized cohort of clinically-stable patients with severe
COPD and healthy matched controls. TNF-alpha increased myotube glucose uptake
and lactate production and enhanced the activity and expression levels of
multiple effectors of muscle glycolytic metabolism in a NF-kappaB-dependent
manner. In addition, TNF-alpha activated HIF signaling, which required classical
NF-kappaB activation. Moreover, knock-down of HIF-1alpha largely attenuated
TNF-alpha-induced increases in glycolytic metabolism. Accordingly, mRNA levels
of HIF-1alpha and of the HIF-1alpha target gene vascular endothelial growth
factor (VEGF) were increased in muscle biopsies of COPD patients compared to
controls which was most pronounced in patients with high levels of muscle
TNF-alpha. TNF-alpha-induced classical NF-kappaB activation enhances muscle
glycolytic metabolism in a HIF-1alpha dependent manner.
TNF-alpha-Induced NF-kappa B Activation Stimulates Skeletal Muscle Glycolytic
Metabolism Through Activation of HIF-1 alpha - Maastricht University Research
Portal
https://cris.maastrichtuniversity.nl/portal/en/publications/tnfalphainduced-nfkappa-b-activation-stimulates-skeletal-muscle-glycolytic-metabolism-through-activation-of-hif1-alpha(c659b0b9-6c50-4cc3-afbe-07c551ddbd40).html
TNF alpha induces expression of HIF-1 mRNA and protein but ...
https://core.ac.uk/display/132816358
TNF alpha induces expression of HIF-1 mRNA and protein but inhibits hypoxic
stimulation of HIF-1 alpha transcriptional activity in airway smooth muscle
cells By S. Tsapournioti, I. Mylonis, A. Hatziefthimiou, M. G. Ioannou, R.
Stamatiou, G. K. Koukoulis, G. Simos, P. A. Molyvdas and E. Paraskeva
TNFα诱导气道平滑肌细胞中HIF-1 mRNA和蛋白的表达,但抑制缺氧刺激HIF-1 alpha转录活性
TNF alpha induces expression of HIF-1 mRNA and protein but inhibits hypoxic
stimulation of HIF-1 alpha transcriptional activity in airway smooth muscle
cells
Provided by: University of Thessaly Institutional Repository
抽象
气道平滑肌细胞(ASMC)参与气道炎性疾病(如哮喘)的组织重塑。炎症和缺氧途径经常相互联系,并且近来已证明低氧诱导因子HIF-1的调节亚基是由细胞因子诱导的。在这里,我们调查与炎症介质TNF和/或低氧对ASMC的单独或联合治疗对HIF-1,HIF-1靶标和炎症标志物表达的影响。在常氧和低氧条件下,TNF均可增强HIF-1蛋白和mRNA水平。
TNF介导的HIF-1基因转录诱导被抑制NF-B通路抑制。
尽管HIF-1蛋白上调,但在常氧下存在TNF的情况下,HIF-1靶基因的转录仍然很低,而在低氧时甚至降低。我们显示,TNF降低HIF-1转录活性是由于抑制HIF-1与ARNT的相互作用并随后阻断了其与HRE的结合。缺氧和TNF对炎症标志物表达的影响之间的比较显示出显着差异:缺氧上调IL-6的表达,而不是RANTES或ICAM的表达,并减少TNF对VCAM的诱导。最后,用TNF进行兔气管条的离体处理可增加缺氧条件下HIF-1蛋白的水平,但会降低HIF-1靶标的表达。总体而言,TNF通过NF-B依赖性途径诱导HIF-1 mRNA合成,但抑制HIF-1与ARNT和DNA的结合,而低氧和TNF对ASMC炎症基因表达具有明显的影响。 J.细胞。生理学。 228:17451753,2013。(c)2013 Wiley Periodicals,Inc
TNF alpha induces expression of HIF-1 mRNA and protein but inhibits hypoxic
stimulation of HIF-1 alpha transcriptional activity in airway smooth muscle
cells
Abstract
Airway smooth muscle cells (ASMCs) participate in tissue remodeling
characteristic of airway inflammatory diseases like asthma. Inflammation and
hypoxia pathways are often interconnected and the regulatory subunit of the
hypoxia inducible factor, HIF-1, has been recently shown to be induced by
cytokines. Here we investigate the effect of individual or combined treatment of
ASMCs with the inflammatory mediator TNF and/or hypoxia on the expression of
HIF-1, HIF-1 targets and inflammation markers. TNF enhances HIF-1 protein and
mRNA levels, under both normoxia and hypoxia. TNF-mediated induction of HIF-1
gene transcription is repressed by inhibition of the NF-B pathway. Despite the
up-regulation of HIF-1 protein, the transcription of HIF-1 target genes remains
low in the presence of TNF at normoxia and is even reduced at hypoxia. We show
that the reduction in HIF-1 transcriptional activity by TNF is due to inhibition
of the interaction of HIF-1 with ARNT and subsequent blocking of its binding to
HREs. Comparison between hypoxia and TNF for their effects on the expression of
inflammatory markers shows significant differences: hypoxia up-regulates the
expression of IL-6, but not RANTES or ICAM, and reduces the induction of VCAM by
TNF. Finally, ex vivo treatment of rabbit trachea strips with TNF increases
HIF-1 protein levels, but reduces the expression of HIF-1 targets under hypoxia.
Overall, TNF induces HIF-1 mRNA synthesis via an NF-B dependent pathway but
inhibits binding of HIF-1 to ARNT and DNA, while hypoxia and TNF have distinct
effects on ASMC inflammatory gene expression. J. Cell. Physiol. 228: 17451753,
2013. (c) 2013 Wiley Periodicals, Inc
TNF alpha induces expression of HIF-1 mRNA and protein but inhibits hypoxic
stimulation of HIF-1 alpha transcriptional activity in airway smooth muscle
cells - CORE
https://core.ac.uk/display/132816358
DHA blocks TPA-induced cell invasion by inhibiting MMP-9 ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5245160
Nov 11, 2016 · In conclusion, DHA is a potent inhibitor of TPA-induced MMP-9
expression, and blocks breast carcinoma cell invasion by modulating the NF-κB
signaling pathway via PPAR-γ upregulation. These findings suggest that DHA may
be an effective therapeutic agent for preventing breast tumor invasion and
metastasis.
DHA通过抑制MCF-7细胞中的PPAR-γ/NF-κB途径来抑制MMP-9表达,从而阻止TPA诱导的细胞侵袭
DHA blocks TPA-induced cell invasion by inhibiting MMP-9
expression via suppression of the PPAR-γ/NF-κB pathway in MCF-7 cells
摘要
二十二碳六烯酸(DHA)是一种omega-3脂肪酸,被认为可用于癌症的预防和治疗。
DHA对癌症转移的有益作用已得到公认;然而,这些作用在乳腺癌中的潜在机制尚不清楚。
细胞侵袭对于肿瘤转移至关重要,并且涉及基质金属蛋白酶(MMP)-9对细胞外基质的降解。
本研究调查了DHA对MCF-7乳腺癌细胞系中12-O-十四烷酰phorbol-13-乙酸盐(TPA)诱导的MMP-9表达和细胞侵袭的抑制作用。 DHA抑制TPA诱导的丝裂原活化蛋白激酶(MAPK)的活化和核因子(NF)-κB的转录,但不抑制活化蛋白1的转录。 DHA增加了过氧化物酶体增殖物激活受体(PPAR)-γ的活性,这种作用可通过应用PPAR-γ拮抗剂GW9662来逆转。此外,GW9662和DHA的联合处理可增加NF-κB相关蛋白的表达。
这些结果表明,DHA通过调节MAPK信号通路和PPAR-γ/NF-κB活性来调节MMP-9表达和细胞侵袭。这表明DHA可能是预防乳腺癌转移的潜在治疗剂。
关键字:DHA,MMP-9,PPAR-γ,侵袭,NF-κB,MCF-7
DHA blocks TPA-induced cell invasion by inhibiting MMP-9 expression via
suppression of the PPAR-γ/NF-κB pathway in MCF-7 cells
Abstract
Docosahexaenoic acid (DHA) is an omega-3 fatty acid that is considered to have
applications in cancer prevention and treatment. The beneficial effects of DHA
against cancer metastasis are well established; however, the mechanisms
underlying these effects in breast cancer are not clear. Cell invasion is
critical for neoplastic metastasis, and involves the degradation of the
extracellular matrix by matrix metalloproteinase (MMP)-9. The present study
investigated the inhibitory effect of DHA on MMP-9 expression and cell invasion
induced by 12-O-tetradecanoylphorbol-13-acetate (TPA) in the MCF-7 breast cancer
cell line. DHA inhibited the TPA-induced activation of mitogen-activated protein
kinase (MAPK) and the transcription of nuclear factor (NF)-κB, but did not
inhibit the transcription of activator protein-1. DHA increased the activity of
peroxisome proliferator-activated receptor (PPAR)-γ, an effect that was reversed
by the application of the PPAR-γ antagonist GW9662. In addition, combined
treatment with GW9662 and DHA increased NF-κB-related protein expression. These
results indicate that DHA regulates MMP-9 expression and cell invasion via
modulation of the MAPK signaling pathway and PPAR-γ/NF-κB activity. This
suggests that DHA could be a potential therapeutic agent for the prevention of
breast cancer metastasis.
Keywords: DHA, MMP-9, PPAR-γ, invasion, NF-κB, MCF-7
DHA blocks TPA-induced cell invasion by inhibiting MMP-9 expression via
suppression of the PPAR-γ/NF-κB pathway in MCF-7 cells
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5245160/
DHA通过TLR‑4 / PPAR‑α途径诱导人恶性乳腺癌组织的凋亡
DHA induces apoptosis of human malignant breast cancer tissues by the
TLR‑4/PPAR‑α pathways
抽象
二十二碳六烯酸(DHA)油是人体重要的多不饱和脂肪酸。有证据表明,DHA对抑制乳腺癌变有益。但是,DHA介导细胞凋亡诱导的机制尚未完全阐明。因此,在本研究中,测定了经DHA油处理的人的总超氧化物歧化酶(t-SOD),过氧化氢酶(CAT),谷胱甘肽过氧化物酶(GSH-PX)和丙二醛(MDA)的活性水平恶性乳房组织。结果表明,与对照相比,DHA显着提高了主要的抗氧化酶水平,包括t-SOD,CAT和GSH-PX,但降低了DHA油处理过的乳腺癌组织中的MDA浓度。此外,DHA显着增加了环(c)AMP
/
cGMP水平的比率,并促进了Toll样受体4(TLR‑4)和过氧化物酶体增殖物激活的受体(PPAR)‑α的表达,因此DHA诱导了乳腺癌细胞凋亡。我们假设TLR‑4和PPAR‑α的水平与DHA在乳腺癌中的抗肿瘤发生特性有关。本研究结果对乳腺癌DHA油的进一步临床开发具有重要意义。
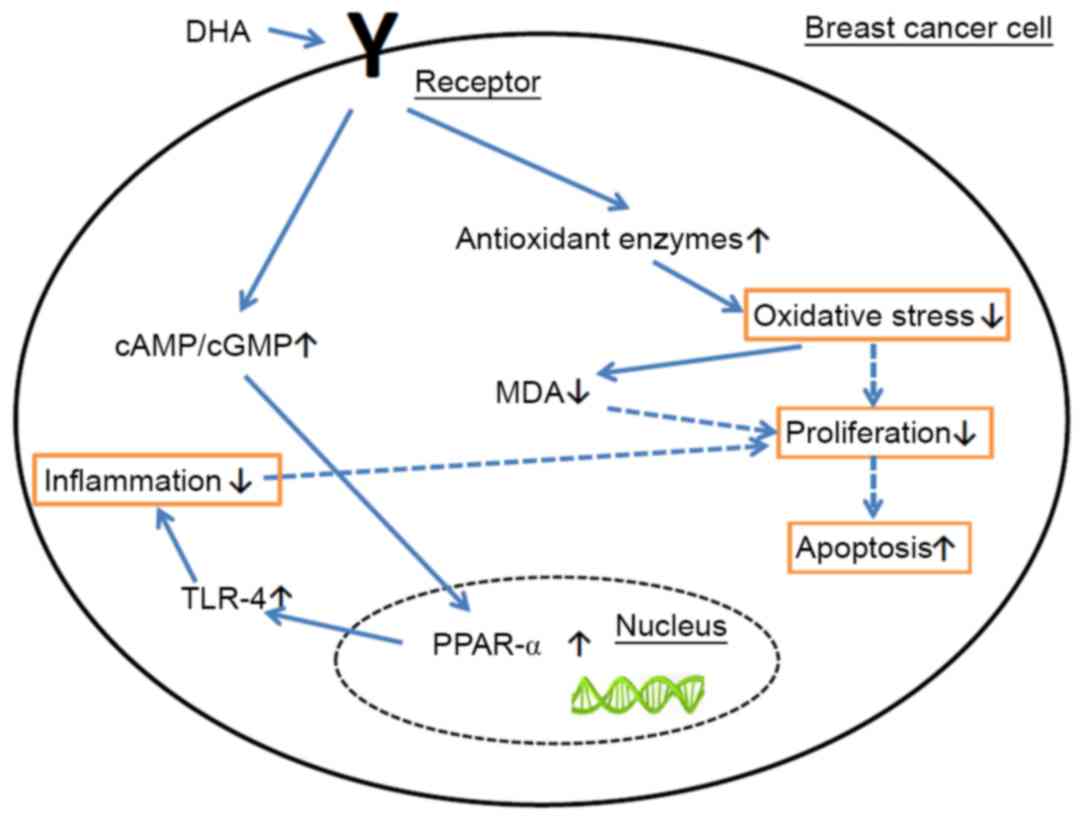
DHA induces apoptosis of human malignant breast cancer tissues by the
TLR‑4/PPAR‑α pathways
Abstract
Docosahexaenoic acid (DHA) oil is an important polyunsaturated fatty acid for
the human body. Evidence has demonstrated that DHA is beneficial for inhibiting
mammary carcinogenesis. However, the mechanisms of DHA mediating apoptosis
induction have not been fully elucidated. Thus, in the present study, the
activity levels of total‑superoxide dismutase (t‑SOD), catalase (CAT),
glutathione‑peroxidase (GSH‑PX) and the concentration of malondialdehyde (MDA)
were determined in DHA oil‑treated human malignant breast tissues. The results
revealed that compared with control, DHA significantly increased the main
antioxidant enzymes levels, including t‑SOD, CAT, and GSH‑PX, but decreased the
MDA concentration in the DHA oil treated breast cancer tissues. Furthermore, DHA
significantly increased the ratio of cyclic (c)AMP/cGMP levels and promoted the
expression of Toll‑like receptor 4 (TLR‑4) and peroxisome proliferator activated
receptor (PPAR)‑α, thus DHA induced breast cancer cell apoptosis. We
hypothesized that the levels of TLR‑4 and PPAR‑α are involved in the
antitumorigenesis properties of DHA in breast cancer. The results of the present
study hold significance for the further clinical development of DHA oil in
breast cancer
DHA induces apoptosis of human malignant breast cancer tissues by the
TLR‑4/PPAR‑α pathways
https://www.spandidos-publications.com/ol/15/3/2967
二十二碳六烯酸(DHA)和多酚通过激活蛋白磷酸酶协同诱导乳腺癌细胞凋亡
Docosahexaenoic acid (DHA) and polyphenols synergistically
induce apoptosis in breast cancer cells by activating protein phosphatases
摘要
我们已经研究了omega-3脂肪酸与饮食多酚结合诱导乳腺癌细胞凋亡的能力。
本研究的目的是研究蛋白质酪氨酸介导的和蛋白质丝氨酸/苏氨酸激酶介导的途径,以评估这些分子的抗癌作用。
在不存在或存在DHA(25μM)的情况下,以各种浓度的多酚(姜黄素,白藜芦醇,杨梅素,槲皮素,绿茶多酚和葡萄皮提取物)处理MDA-MB-231细胞。使用WST-1和LDH分析测定对细胞活力的影响。使用充满活力的凋亡分析试剂盒分析凋亡的诱导。使用磷酸丝氨酸和磷酸酪氨酸特异性抗体分析细胞蛋白的磷酸化。
这些研究结果表明,多酚与DHA协同作用,诱导乳腺癌细胞凋亡。
测试的所有多酚和omega-3脂肪酸均以剂量依赖的方式抑制乳腺癌细胞中几种蛋白质的酪氨酸和丝氨酸/苏氨酸磷酸化。但是,这些试剂刺激了38 kDa的蛋白质的丝氨酸磷酸化,蛋白质经Western分析鉴定为p38激酶,是MAP激酶超家族的成员。我们的数据表明,饮食中的多酚和omega-3脂肪酸可以通过激活细胞蛋白磷酸酶来抑制乳腺癌的生长,磷酸化酶会诱导p38激酶(参与细胞凋亡的调控蛋白)中Thr 180 / Tyr 182残基的下游磷酸化。
Docosahexaenoic acid (DHA) and polyphenols synergistically induce apoptosis in breast cancer cells by activating protein phosphatases
Methodist Research Institute, 1800 N Capitol Ave, Indianapolis, IN, 46202
Abstract We have investigated the ability of omega-3 fatty acids combined
with dietary polyphenols to induce apoptosis in breast cancer cells. The
objective of the present study was to investigate the protein tyrosine-mediated
and protein serine/threonine kinase-mediated pathways to assess the anticancer
effects of these molecules. MDA-MB-231 cells were treated with various
concentrations of polyphenols (Curcumin, Resveratrol, Myricetin, Quercetin,
Green Tea Polyphenols and Grape Skin Extracts) in the absence or presence of of
DHA (25 μM). Effects on cell viability were measured using WST-1 and LDH assays.
Induction of apoptosis was analyzed using a vibrant apoptotic assay kit.
Phosphorylation of cellular proteins was analyzed using phosphoserine- and
phosphotyrosine-specific antibodies. Results of these studies demonstrate that
polyphenols act synergistically with DHA to induce apoptosis in breast cancer
cells. All of the polyphenols and omega-3 fatty acids tested inhibited tyrosine
and serine/threonine phosphorylation of several proteins in breast cancer cells
in a dose-dependent manner. However, these agents stimulated serine
phosphorylation of a protein at 38 kDa, which was identified using Western
analysis as p38 kinase, a member of MAP-kinase super-family. Our data indicate
that dietary polyphenols and omega-3 fatty acids may inhibit breast cancer
growth by activating cellular protein phosphatases, which induces downstream
phosphorylation of Thr 180/Tyr 182 residues on p38 kinase, a regulatory protein
involved in cell apoptosis.
Docosahexaenoic acid (DHA) and polyphenols synergistically induce apoptosis
in breast cancer cells by activating protein phosphatases | The FASEB Journal
https://www.fasebj.org/doi/abs/10.1096/fasebj.20.4.A567
癌症中的NF-κB:生死攸关的问题
NF-κB in cancer: A Matter of Life and Death
Bharat B.Aggarwal和Bokyung Sung
德州大学医学博士Anderson癌症中心,实验治疗学系细胞因子研究实验室,德克萨斯州休斯敦
摘要
NF-κB的激活与癌症中的各种细胞过程有关,包括炎症,转化,增殖,血管生成,侵袭,转移,化学抗性和放射抗性。尽管急性炎症会介导先天和体液免疫,但慢性炎症与肿瘤发生有关。因此,抑制NF-κB在使肿瘤对化学治疗剂敏感方面具有治疗潜力。然而,NF-κB的普遍抑制可导致严重的宿主毒性,而对肿瘤的影响最小。
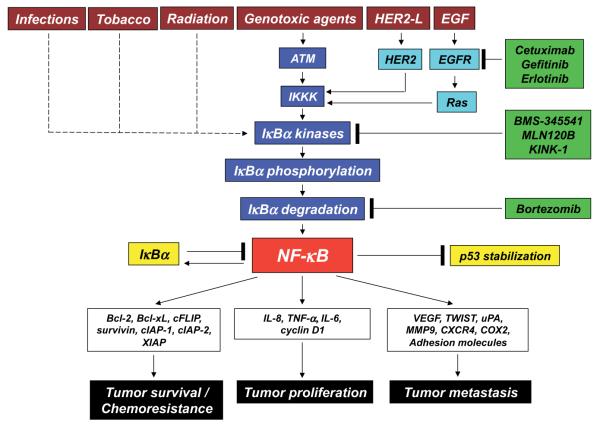
图1
导致肿瘤细胞中NF-κB活化的途径以及随后NF-κB活化在肿瘤发生中的作用。 ATM,共济失调-毛细血管扩张突变; EGF,表皮生长因子;
EGFR,表皮生长因子受体; HER2-L,HER2配体。
NF-κB in cancer: A Matter of Life and Death
Bharat B. Aggarwal and Bokyung Sung
Cytokine Research Laboratory, Department of Experimental Therapeutics,
University of Texas MD Anderson Cancer Center, Houston, Texas
Summary
Activation of NF-κB has been linked to various cellular processes in cancer,
including inflammation, transformation, proliferation, angiogenesis, invasion,
metastasis, chemoresistance, and radioresistance. Although acute inflammation
mediates innate and humoral immunity, chronic inflammation has been linked to
tumorigenesis. Thus, inhibition of NF-κB has therapeutic potential in
sensitization of tumors to chemotherapeutic agents; however, generalized
suppression of NF-κB can result in serious host toxicity with minimum effect on
the tumor.
NF-κB in cancer: A Matter of Life and Death
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3392037/
NF-kB被认为是一种“生物开关”在炎症和癌症中发挥至关重要的作用
NF-κB[核因子-κB]通过诱导炎症反应而在炎症中起重要作用。
NF-κB的活性与动脉粥样硬化,关节炎,炎性肠病,类风湿性关节炎和败血性休克,恶性转化和肿瘤生长(例如某些血液癌和实体瘤)以及骨分解和重建(骨质疏松症)有关。
NF-κB通常被认为是一种“生物开关”,可以在细胞中关闭以治疗这些疾病。
“NF-κB被多种因子激活,包括细胞因子,氧化剂自由基,吸入颗粒,紫外线辐射以及细菌或病毒产物。
NF-κB [Nuclear factor-kappaBeta] plays a central role in inflammation through
its ability to induce pro-inflammatory responses.
The activity of NF-κB has been implicated in atherosclerosis, arthritis,
inflammatory bowel disease, rheumatoid arthritis, and septic shock, malignant
transformation and tumour growth (e.g., certain blood cancers and solid
tumours), and bone breakdown and rebuilding (osteoporosis).
NF-κ B can be generally thought of as a "biological switch" that can be turned
off in cells to treat these diseases.
“NF-κB is activated by several agents, including cytokines, oxidant free
radicals, inhaled particles, ultraviolet irradiation, and bacterial or viral
products.
Positive Health Online | Article - Curcumin vs Cancer
http://www.positivehealth.com/article/cancer/curcumin-vs-cancer
NFκB信号通路在癌症中的作用
Role of the NFκB-signaling pathway in cancer
夏龙正,#1,*谭世明,#1,*周玉娟,1景冠,1王兰,1琳达·奥阳,1田宇彤,1陆柳,1民苏,1王辉,1曹德良,1 ,2和钱千金1
作者信息版权和许可信息免责声明
1湖南省湘雅医学院湖南省肿瘤医院和附属肿瘤医院湖南省转化放射肿瘤学重点实验室,湖南长沙
2美国伊利诺伊州斯普林菲尔德市南伊利诺伊大学医学院西蒙斯癌症研究所医学微生物学,免疫学和细胞生物学系
#平均贡献。
通讯:廖千金;曹德良,中南大学湘雅医学院湖南省肿瘤医院和附属肿瘤医院湖南省转化放射肿瘤学重点实验室,湖南长沙桐子坡路283号,湖南长沙410013,电话+86
731 8865
抽象
癌症是一群恶性生长且不受控制地增殖的细胞。目前,癌症的治疗方式主要包括手术,化学疗法,放射疗法,分子靶向疗法,基因疗法和免疫疗法。然而,迄今为止,这些治疗的疗效受到肿瘤的特定特征的限制。信号通路的异常激活参与了肿瘤的发病机理,并在癌症的生长,进展和复发中起着至关重要的作用。针对致癌信号中效应子的靶向疗法已改善了癌症患者的预后。
NFκB是参与癌症的发病机制和治疗的重要信号通路。已经在各种肿瘤组织中证明了NFκB信号传导通路的过度激活,并且针对该信号通路的靶向癌症治疗的研究已经成为热门话题。在这篇综述中,我们更新了对癌症中NFκB信号通路的当前了解。
关键词:核因子κB,p65,信号通路,癌症,炎症
Role of the NFκB-signaling pathway in cancer
Longzheng Xia,#1,* Shiming Tan,#1,* Yujuan Zhou,1 Jingguan Lin,1 Heran Wang,1
Linda Oyang,1 Yutong Tian,1 Lu Liu,1 Min Su,1 Hui Wang,1 Deliang Cao,1,2 and
Qianjin Liao1
Author information Copyright and License information Disclaimer
1Hunan Key Laboratory of Translational Radiation Oncology, Hunan Cancer Hospital
and Affiliated Cancer Hospital of Xiangya School of Medicine, Changsha, Hunan,
China
2Department of Medical Microbiology, Immunology, and Cell Biology, Simmons
Cancer Institute, Southern Illinois University School of Medicine, Springfield,
IL, USA
#Contributed equally.
Correspondence: Qianjin Liao; Deliang Cao, Hunan Key Laboratory of Translational
Radiation Oncology, Hunan Cancer Hospital and Affiliated Cancer Hospital of
Xiangya School of Medicine, Central South University, 283 Tongzipo Road,
Changsha, Hunan 410013, China, Tel +86 731 8865
Abstract
Cancer is a group of cells that malignantly grow and proliferate uncontrollably.
At present, treatment modes for cancer mainly comprise surgery, chemotherapy,
radiotherapy, molecularly targeted therapy, gene therapy, and immunotherapy.
However, the curative effects of these treatments have been limited thus far by
specific characteristics of tumors. Abnormal activation of signaling pathways is
involved in tumor pathogenesis and plays critical roles in growth, progression,
and relapse of cancers. Targeted therapies against effectors in oncogenic
signaling have improved the outcomes of cancer patients. NFκB is an important
signaling pathway involved in pathogenesis and treatment of cancers. Excessive
activation of the NFκB-signaling pathway has been documented in various tumor
tissues, and studies on this signaling pathway for targeted cancer therapy have
become a hot topic. In this review, we update current understanding of the
NFκB-signaling pathway in cancer.
Keywords: nuclear factor kappa-B, p65, signaling pathway, cancer, inflammation
TNF家族细胞因子对NF-κB的调控
Regulation of NF-κB by TNF Family Cytokines
NF-κB家族的可诱导转录因子响应各种刺激而被激活。在最典型的NF-κB诱导剂中,是细胞因子TNF家族的成员。
NF-κB和TNF的研究已经紧密地交织在一起超过25年了。
NF-κB和TNF相互连接的最有说服力的例子可能来自无法激活NF-kB的基因敲除小鼠的分析。此类小鼠在胚胎中死亡,但是,TNF或TNFR1的缺失可以挽救致命性,从而说明NF-κB作为TNF转录反应的关键调节因子的重要作用。
NF-κB与TNF细胞因子之间的生理联系是众多的,并且在针对单个TNF家族成员的文章中得到了最好的探讨。相反,在本综述中,我们探讨了TNF细胞因子信号传导的一般机制,重点是导致分别通过TNFR1和CD40激活所谓的经典和非经典NF-κB途径的上游信号事件。
关键词:NF-κB,TNF,CD40 IKK,TRAF,RIP,信号传导,炎症
Regulation of NF-κB by TNF Family Cytokines
The NF-κB family of inducible transcription factors is activated in response to
a variety of stimuli. Amongst the best-characterized inducers of NF-κB are
members of the TNF family of cytokines. Research on NF-κB and TNF have been
tightly intertwined for more than 25 years. Perhaps the most compelling examples
of the interconnectedness of NF-κB and the TNF have come from analysis of
knock-out mice that are unable to activate NF-kB. Such mice die embryonically,
however, deletion of TNF or TNFR1 can rescue the lethality thereby illustrating
the important role of NF-κB as the key regulator of transcriptional responses to
TNF. The physiological connections between NF-κB and TNF cytokines are numerous
and best explored in articles focusing on a single TNF family member. Instead,
in this review, we explore general mechanisms of TNF cytokine signaling, with a
focus on the upstream signaling events leading to activation of the socalled
canonical and noncanonical NF-κB pathways by TNFR1 and CD40 respectively.
Keywords: NF-kappaB, TNF, CD40 IKK, TRAF, RIP, signaling, inflammation
Regulation of NF-κB by TNF Family Cytokines
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4156877/
维生素C诱导的乳腺癌细胞死亡的机制:含半胱氨酸蛋白的氧化还原修饰,细胞周期停滞和翻译抑制
Redox modifications of cysteine-containing proteins, cell cycle arrest and translation inhibition: Involvement in vitamin C-induced breast cancer cell death
a Institut Curie, PSL Research University, CNRS UMR 3348, Université
Paris-Sud, Université Paris-Saclay, Orsay, France
b Institut Jacques Monod, CNRS UMR 7592, Mass Spectrometry Laboratory,
Université Paris Diderot, Paris, France
c Institut Gustave Roussy, CNRS UMR 8200, Université Paris-Sud, Villejuif,
France
强调
•抗坏血酸(AA)在体外对乳腺癌细胞显示出高细胞毒性。
•H2O2是AA对乳腺癌细胞毒性的主要介质。
•AA处理会改变大量含半胱氨酸的蛋白质的氧化还原状态。
•AA处理触发细胞周期停滞和翻译抑制。
•PRDX1表达水平可能是细胞对AA反应的预测生物标志物。
抽象
维生素C(VitC)在高药理浓度下具有促氧化剂特性,有利于将VitC重新用作抗癌治疗剂。但是,还部分了解VitC的基于氧化还原的抗癌特性。我们研究了VitC,抗坏血酸(AA)和脱氢抗坏血酸(DHA)的还原形式和氧化形式之间的区别,即针对乳腺癌细胞的细胞毒性和氧化还原机制。
我们的数据显示,AA在体外对三阴性乳腺癌(TNBC)细胞系显示出比DHA高的细胞毒性。 AA对非TNBC细胞表现出相似的细胞毒性,而对非癌细胞则只有很小的有害作用。
使用MDA-MB-231,一种典型的TNBC细胞系,我们观察到AA和DHA诱导的细胞毒性与细胞氧化还原状态的改变有关。过氧化氢(H2O2)积累在细胞外培养基中和不同的细胞内区室中,细胞内的谷胱甘肽氧化程度较小,在AA诱导的细胞毒性中起关键作用。相反,DHA影响谷胱甘肽氧化,细胞毒性较小。 “氧化还原酶”方法显示,AA处理改变了关键抗氧化剂和许多含半胱氨酸的蛋白质的氧化还原状态,其中包括许多核酸结合蛋白以及涉及RNA和DNA代谢以及高能过程的蛋白质。我们表明,细胞周期停滞和翻译抑制与AA诱导的细胞毒性有关。最后,生物信息学分析和生物学实验确定了过氧化物酶1(PRDX1)的表达水平与乳腺癌细胞中AA的不同细胞毒性相关,提示PRDX1的潜在预测价值。
这项研究提供了对基于氧化还原的VitC抗癌活性机制的见解,表明VitC和基于VitC的合理药物组合的药理剂量可能是三阴性乳腺癌的新型治疗机会。
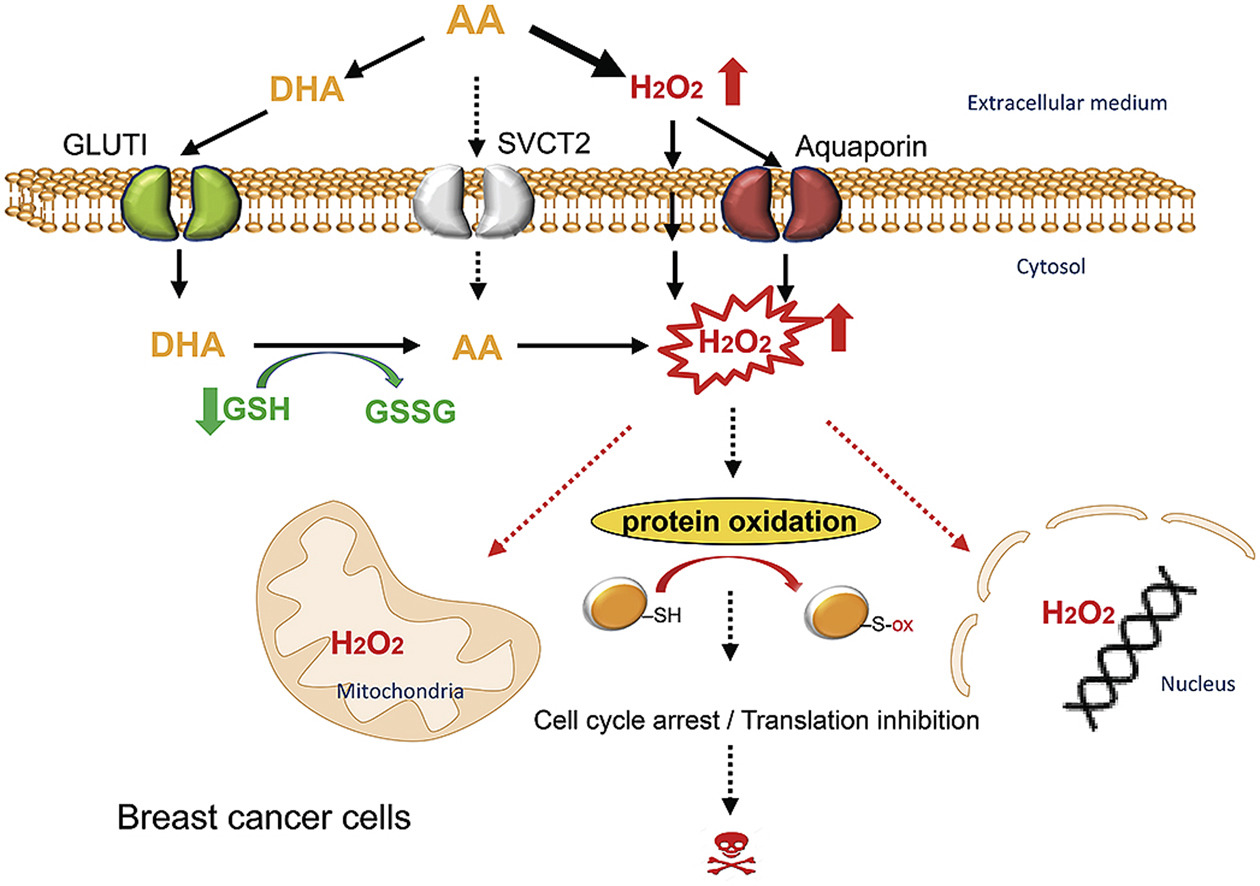
Redox modifications of cysteine-containing proteins, cell cycle arrest and
translation inhibition: Involvement in vitamin C-induced breast cancer cell
death
Highlights
• Ascorbic acid (AA) displays high cytotoxicity towards breast cancer cells in
vitro.
• H2O2 is the main mediator of AA cytotoxicity toward breast cancer cells.
• AA treatment alters the redox state of large number of cysteine-containing
proteins.
• AA treatment triggers cell cycle arrest and translation inhibition.
• PRDX1 expression levels could be a predictive biomarker for cellular response
to AA.
Abstract
Vitamin C (VitC) possesses pro-oxidant properties at high pharmacologic
concentrations which favor repurposing VitC as an anti-cancer therapeutic agent.
However, redox-based anticancer properties of VitC are yet partially understood.
We examined the difference between the reduced and oxidized forms of VitC,
ascorbic acid (AA) and dehydroascorbic acid (DHA), in terms of cytotoxicity and
redox mechanisms toward breast cancer cells. Our data showed that AA displayed
higher cytotoxicity towards triple-negative breast cancer (TNBC) cell lines in
vitro than DHA. AA exhibited a similar cytotoxicity on non-TNBC cells, while
only a minor detrimental effect on noncancerous cells. Using MDA-MB-231, a
representative TNBC cell line, we observed that AA- and DHA-induced cytotoxicity
were linked to cellular redox-state alterations. Hydrogen peroxide (H2O2)
accumulation in the extracellular medium and in different intracellular
compartments, and to a lesser degree, intracellular glutathione oxidation,
played a key role in AA-induced cytotoxicity. In contrast, DHA affected
glutathione oxidation and had less cytotoxicity. A “redoxome” approach revealed
that AA treatment altered the redox state of key antioxidants and a number of
cysteine-containing proteins including many nucleic acid binding proteins and
proteins involved in RNA and DNA metabolisms and in energetic processes. We
showed that cell cycle arrest and translation inhibition were associated with
AA-induced cytotoxicity. Finally, bioinformatics analysis and biological
experiments identified that peroxiredoxin 1 (PRDX1) expression levels correlated
with AA differential cytotoxicity in breast cancer cells, suggesting a potential
predictive value of PRDX1. This study provides insight into the redox-based
mechanisms of VitC anticancer activity, indicating that pharmacologic doses of
VitC and VitC-based rational drug combinations could be novel therapeutic
opportunities for triple-negative breast cancer.
Redox modifications of cysteine-containing proteins, cell cycle arrest and
translation inhibition: Involvement in vitamin C-induced breast cancer cell
death - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S2213231719307499
Researchers in Japan report extracellular iron (outside cancer cells)
decomposes hydrogen peroxide generated by vitamin C. The inhibition of cancer
cell growth via vitamin C (hydrogen peroxide) is completely blocked by the
presence of iron. [Scientific Reports 2018] This was demonstrated in leukemia
cells (cancer of the blood) in a lab dish.
Depletion of Iron From Tumor Cells Prior To Vitamin C ...
knowledgeofhealth.com/depletion-iron-from-tumor-%e2%80%a8prior-vitamin-c-%e2%80%a8quell…
Depletion of Iron From Tumor Cells Prior To Vitamin C ...
https://www.lewrockwell.com/2019/01/bill-sardi/depletion-of-iron-from-tumor-cells...
Jan 02, 2019 · Researchers in Japan report extracellular iron (outside cancer
cells) decomposes hydrogen peroxide generated by vitamin C. The inhibition of
cancer cell growth via vitamin C (hydrogen peroxide) is completely blocked by
the presence of iron. [Scientific Reports 2018] This was demonstrated in
leukemia cells (cancer of the blood) in a lab dish. Given that most of the iron
stored in the body is …
2008年8月4日,星期一
美国国立卫生研究院:维生素C注射减慢小鼠肿瘤的生长
Vitamin C Injections Slow Tumor Growth in Mice
美国国立卫生研究院(NIH)的研究人员在报告中指出,大剂量注射维生素C(也称为抗坏血酸或抗坏血酸)可在脑癌,卵巢癌和胰腺癌的小鼠模型中将肿瘤重量和生长率降低约50%。
2008年8月5日出版的《美国国家科学院院刊》。研究人员将抗坏血酸的抗癌作用追溯到肿瘤周围细胞外液中过氧化氢的形成。正常细胞不受影响。
口服时,自然的生理控制可以精确地调节人体吸收的抗坏血酸的量。
“当您每天吃含有200毫克维生素C的食物(例如2个橙子和一份西兰花)时,您的身体可防止抗坏血酸的血液含量超出狭窄范围,”医学博士Mark
Levine说道。美国国立卫生研究院(NIH)糖尿病与消化和肾脏疾病研究所(NIDDK)的分子和临床营养科主任和负责人。为了绕过这些正常控制,NIH科学家将抗坏血酸注射到患有侵袭性脑,卵巢和胰腺肿瘤的啮齿动物的静脉或腹腔中。通过这样做,他们能够提供高剂量的抗坏血酸,每天每公斤体重最多4克。莱文说:“在这些高剂量下,我们希望看到类似药物的活性可能对癌症治疗有用。”
维生素C对健康至关重要,长期缺乏维生素会导致坏血病,甚至导致死亡。一些具有重要生化功能的称为酶的蛋白质需要维生素正常工作。维生素C还可以充当抗氧化剂,保护细胞免受自由基的破坏。但是,NIH研究人员测试了抗坏血酸的想法,当高剂量注射时,抗坏血酸可能具有抗氧化剂而不是抗氧化剂活性。科学家推测,前氧化剂会产生自由基并形成过氧化氢,可能杀死肿瘤细胞。在针对43种癌症和5种正常细胞系的实验室实验中,研究人员发现,高浓度的抗坏血酸在75%的测试癌细胞系中具有抗癌作用,而保留了正常细胞。研究人员在他们的论文中还表明,这些高抗坏血酸浓度可以在人体内实现。
然后,研究小组对免疫缺陷小鼠中快速扩散的卵巢,胰腺和成胶质细胞瘤(脑)肿瘤的抗坏血酸注射剂进行了测试。注射抗坏血酸可使肿瘤生长和重量减少41%至53%。在30%的胶质母细胞瘤对照中,癌症已经扩散到其他器官,但是抗坏血酸处理过的动物没有癌症扩散的迹象。研究人员总结说:“这些临床前数据为在人类癌症治疗中推进药理学抗坏血酸提供了坚实的基础。”
大约30年前,当病例系列数据显示可能有益时,人们对维生素C作为一种潜在的癌症治疗方法的兴趣达到顶峰。但是,在1979年和1985年,其他研究人员在两项双盲,安慰剂对照的临床试验中报告,口服高剂量维生素C的癌症患者无益。
多项观察结果促使NIH研究人员重新将抗坏血酸作为一种癌症疗法。
“在过去的十二年中进行的临床和药代动力学研究表明,血浆和组织中的口服抗坏血酸水平受到严格控制。在这种情况下,口服和静脉内给予抗坏血酸,但在试验中仅口服给予抗坏血酸。
“只有注射抗坏血酸的药物才能达到抗肿瘤作用所需的浓度。”莱文说,他指出,抗坏血酸作为一种癌症治疗药物的新临床试验尚处于计划阶段。
Levine对饮食中维生素C的调节和吸收的早期研究数据被用于2000年美国医学研究所对维生素的推荐饮食津贴的修订中。在当前研究中,Levine领导了NIDDK和美国国家医学会的科学家团队美国国立卫生研究院(NIH)和堪萨斯大学的癌症研究所(NCI)。他说:“美国国立卫生研究院独特的翻译环境使研究人员可以从事具有高潜力的智力高风险,开箱即用的思维,使我们得以从事这项工作。”
有关癌症的更多信息,请访问NCI网站www.cancer.gov,或致电1-800-4-CANCER(1-800-422-6237)致电NCI癌症信息服务。
NIDDK开展并支持糖尿病以及其他内分泌和代谢性疾病的研究;消化系统疾病,营养和肥胖;以及肾脏,泌尿科和血液科疾病。这些疾病涉及所有年龄段和种族的医学,并且困扰着各个年龄段和各个民族的人们,这些疾病涵盖了影响美国人的一些最常见,最严重和致残的疾病。有关NIDDK及其程序的更多信息,请访问www.niddk.nih.gov。
关于美国国立卫生研究院(NIH):美国国立卫生研究院(NIH)是美国的医学研究机构,包括27个研究所和中心,并且是美国卫生与公共服务部的一部分。
NIH是进行和支持基础,临床和转化医学研究的主要联邦机构,并且正在调查常见和罕见疾病的病因,治疗方法和治愈方法。有关NIH及其计划的更多信息,请访问www.nih.gov。
Monday, August 4, 2008
Vitamin C Injections Slow Tumor Growth in Mice
High-dose injections of vitamin C, also known as ascorbate or ascorbic acid,
reduced tumor weight and growth rate by about 50 percent in mouse models of
brain, ovarian, and pancreatic cancers, researchers from the National Institutes
of Health (NIH) report in the August 5, 2008, issue of the Proceedings of the
National Academy of Sciences. The researchers traced ascorbate’s anti-cancer
effect to the formation of hydrogen peroxide in the extracellular fluid
surrounding the tumors. Normal cells were unaffected.
Natural physiologic controls precisely regulate the amount of ascorbate absorbed
by the body when it is taken orally. "When you eat foods containing more than
200 milligrams of vitamin C a day — for example, 2 oranges and a serving of
broccoli — your body prevents blood levels of ascorbate from exceeding a narrow
range," says Mark Levine, M.D., the study’s lead author and chief of the
Molecular and Clinical Nutrition Section of the National Institute of Diabetes
and Digestive and Kidney Diseases (NIDDK), part of the NIH. To bypass these
normal controls, NIH scientists injected ascorbate into the veins or abdominal
cavities of rodents with aggressive brain, ovarian, and pancreatic tumors. By
doing so, they were able to deliver high doses of ascorbate, up to 4 grams per
kilogram of body weight daily. "At these high injected doses, we hoped to see
drug-like activity that might be useful in cancer treatment," said Levine.
Vitamin C plays a critical role in health, and a prolonged deficiency leads to
scurvy and eventually to death. Some proteins known as enzymes, which have vital
biochemical functions, require the vitamin to work properly. Vitamin C may
also act as an antioxidant, protecting cells from the damaging effects of free
radicals. The NIH researchers, however, tested the idea that ascorbate, when
injected at high doses, may have prooxidant instead of antioxidant activity.
Prooxidants would generate free radicals and the formation of
hydrogen peroxide(H2O2), which, the scientists hypothesized, might kill
tumor cells. In their laboratory experiments on 43 cancer and 5 normal cell
lines, the researchers discovered that high concentrations of ascorbate had
anticancer effects in 75 percent of cancer cell lines tested, while sparing
normal cells. In their paper, the researchers also showed that these high
ascorbate concentrations could be achieved in people.
The team then tested ascorbate injections in immune-deficient mice with rapidly
spreading ovarian, pancreatic, and glioblastoma (brain) tumors. The ascorbate
injections reduced tumor growth and weight by 41 to 53 percent. In 30 percent of
glioblastoma controls, the cancer had spread to other organs, but the
ascorbate-treated animals had no signs of disseminated cancer. "These
pre-clinical data provide the first firm basis for advancing pharmacologic
ascorbate in cancer treatment in humans," the researchers conclude.
Interest in vitamin C as a potential cancer therapy peaked about 30 years ago
when case series data showed a possible benefit. In 1979 and 1985, however,
other researchers reported no benefit for cancer patients taking high oral doses
of vitamin C in two double-blind, placebo-controlled clinical trials.
Several observations led the NIH researchers to revisit ascorbate as a cancer
therapy. "Clinical and pharmacokinetic studies conducted in the past 12 years
showed that oral ascorbate levels in plasma and tissue are tightly controlled.
In the case series, ascorbate was given orally and intravenously, but in the
trials ascorbate was just given orally. It was not realized at the time that
only injected ascorbate might deliver the concentrations needed to see an
anti-tumor effect," said Levine, who noted that new clinical trials of ascorbate
as a cancer treatment are in the planning stages.
Data from Levine’s earlier studies of the regulation and absorption of dietary
vitamin C were used in the revision of the Institute of Medicine’s Recommended
Dietary Allowance for the vitamin in 2000. In the current study, Levine led a
team of scientists from the NIDDK and the National Cancer Institute (NCI), both
components of the NIH, as well as the University of Kansas. "NIH’s unique
translational environment, where researchers can pursue intellectual high-risk,
out-of-the-box thinking with high potential payoff, enabled us to pursue this
work," he said.
For more information about cancer, please visit the NCI website at
www.cancer.gov, or call NCI's Cancer Information Service at 1-800-4-CANCER
(1-800-422-6237).
The NIDDK conducts and supports research in diabetes and other endocrine and
metabolic diseases; digestive diseases, nutrition, and obesity; and kidney,
urologic, and hematologic diseases. Spanning the full spectrum of medicine and
afflicting people of all ages and ethnic groups, these diseases encompass some
of the most common, severe, and disabling conditions affecting Americans. For
more information about NIDDK and its programs, see www.niddk.nih.gov.
About the National Institutes of Health (NIH): NIH, the nation's medical
research agency, includes 27 Institutes and Centers and is a component of the
U.S. Department of Health and Human Services. NIH is the primary federal agency
conducting and supporting basic, clinical, and translational medical research,
and is investigating the causes, treatments, and cures for both common and rare
diseases. For more information about NIH and its programs, visit www.nih.gov.
NIH…Turning Discovery Into Health®
Vitamin C Injections Slow Tumor Growth in Mice, August 4, 2008 News Release -
National Institutes of Health (NIH)
https://www.nih.gov/news-events/news-releases/vitamin-c-injections-slow-tumor-growth-mice
多种多发性骨髓瘤肿瘤细胞可通过药理学上的抗坏血酸
Multiple Myeloma Tumor Cells are Selectively Killed by Pharmacologically-dosed Ascorbic Acid
Jiliang Xia,a,b,1 Hongwei Xu,a,1 Xiaoyan Zhang,a,c,1 Chantal Allamargot,d Kristen L. Coleman,a Randy Nessler,d Ivana Frech,a Guido Tricot,a,⁎,2 and Fenghuang Zhana,⁎,2
美国爱荷华州爱荷华市爱荷华大学霍顿综合癌症中心血液学、肿瘤学和血液与骨髓移植学部医学博士
南方医科大学基础医学院肿瘤研究所,广州,中国
华东理工大学,上海,中国
美国爱荷华州爱荷华市爱荷华大学中央显微镜研究中心
圭多经编针织物:ude.awoiu@tocirt-odiug;凤凰詹:ude.awoiu@nahz-gnauhgnef
⁎相应作者:血液学,肿瘤和血液和骨髓移植,爱荷华大学,585年牛顿Rd,
52242爱荷华市,IA、美国。爱荷华大学血液学、肿瘤学和血液与骨髓移植系。awoiu@tocirt-odiug,
ude.awoiu@nahz-gnauhgnef
夏季良、徐宏伟和张晓燕对本文的贡献相当。
2 drs。特里科特和詹对本文的贡献相当。
EBioMedicine
选择性杀伤。 2017年4月; 18:41-49。
摘要
大剂量化学疗法治疗多发性骨髓瘤(MM)可能会危及生命,因为它对正常细胞具有毒性,因此有必要仅靶向肿瘤细胞和/或降低标准药物剂量而不会失去疗效。
我们表明,在铁的存在下,药理学剂量的抗坏血酸(PAA)导致形成高反应性氧(ROS),导致细胞死亡。 PAA选择性杀死源自MM和阴燃性MM(SMM)的CD138 + MM肿瘤细胞,但不能杀死未明确意义的单克隆丙种球蛋白病(MGUS)患者。 PAA单独或与美法仑合用可抑制MM异种移植小鼠的肿瘤形成。
这项研究显示了PAA在体外和体内对原代癌细胞和细胞系的功效。
关键词:多发性骨髓瘤,药理学上服用的抗坏血酸,细胞凋亡诱导因子1缩写:MM,多发性骨髓瘤; ROS,活性氧; SMM,阴燃MM; IMID,免疫调节药物; ASCT,自体干细胞移植; CR,完全缓解; PAA,药理学剂量的抗坏血酸; LIP,不稳定的铁池; GEP,基因表达谱; BM,骨髓; IVIS,体内成像系统; WT,野生型; DFO,去铁胺; AIF1,细胞凋亡诱导因子1
介绍
1.多发性骨髓瘤(MM)是浆细胞肿瘤。四种活性类别的药物,包括糖皮质激素,DNA烷基化药(美法仑),蛋白酶体抑制剂(硼替佐米和卡非佐米)和免疫调节剂(沙利度胺,来那度胺和泊马利度胺),结合或不结合自体干细胞移植(ASCT)已导致完全缓解( CRs)在大多数新诊断的MM患者中(Alexanian等,2014,Fu等,2013,Terpos等,2014,Wang等,2014,Sonneveld等,2013,Gay等)等人,2013年,Liu等人,2013年,Bergsagel,2014年)。这些治疗极大地改善了患者的无进展生存期和总体生存期。但是,至少有三个主要问题限制了这些药物的给药:1.所有这些药物均靶向肿瘤细胞和非肿瘤细胞; 2.通过将烷基化剂与任何一种免疫调节药物(IMIDs)结合使用,已发现血液学毒性增加(Bergsagel,2014年); 3.高剂量的DNA碱化剂(例如美法仑)对肠道上皮细胞和造血干细胞具有很强的细胞毒性(Shaw等人,2014)。处理高剂量马法兰的非选择性毒性的一种方法是将其与另一种非常特定地靶向肿瘤细胞的药物联合使用,从而降低马法兰的剂量而不损失疗效。
1970年代,卡梅伦和鲍林(Cameron and Pauling)报告说,高剂量的维生素C可提高癌症患者的生存率(Cameron and Pauling,1976; Cameron and Pauling,1978)。最近,有报告显示,以药理学剂量服用抗坏血酸(PAA)50-100 g(Chen等,2008; Padayatty等,2004; Hoffer等,2008; Padayatty等,2006; Welsh等。 (2013年),静脉注射具有有效的抗癌活性,爱荷华大学和其他中心正在研究其作为抗癌治疗的作用(Du等,2012; Ma等,2014)。在存在催化性金属离子(如铁)的情况下,静脉内给予的PAA会发挥促氧化剂作用,导致形成高活性氧(ROS),从而导致细胞死亡(Yun等,2015; Ma等,2014; Du等,2012; Chen等,2007; Chen等,2005)。在先前的研究中,我们报道了MM细胞中的不稳定铁池(labile iron pool, LIP)显着升高(Gu et al。,2015),这表明PAA治疗应相当有选择性地靶向MM细胞。正如我们小组所证实的那样,较高的LIP是唯一已知的哺乳动物细胞铁输出铁蛋白转运蛋白1(Fpn1)在MM中低表达的直接结果。这些发现使我们得出这样的假设:PAA可能特异性地靶向高铁含量的MM细胞,并且可能与常用的MM疗法联合发挥协同作用。
3.3。药理抗坏血酸的治疗效果取决于细胞铁和活性氧的种类
随后我们想知道PAA是否通过产生ROS选择性杀死MM肿瘤细胞,我们用N-乙酰半胱氨酸(NAC)或过氧化氢酶处理OCI-MY5
MM野生型(WT)细胞。过氧化氢酶和NAC均可保护细胞免受氧化损伤。用NAC和过氧化氢酶预处理的OCI-MY5细胞即使在高剂量下也对PAA产生抗性(图2A)。重要的是,在PAA处理之前向OCI-MY5细胞中添加铁螯合剂去铁胺(DFO)也足以防止PAA诱导的细胞死亡(图2A),但选择性铜螯合剂铜代嘌呤二磺酸二钠盐(BCS)却足以防止这种情况。不能阻止MM细胞死亡(补充图5A),表明铁对于PAA实现其抗癌活性至关重要。
DFO是一种膜渗透性差的铁螯合剂,但是,它已被用于多种研究中螯合细胞内铁,包括那些描述表达Fpn1的细胞的研究(Asano等人,2011;
Paradkar等人,2008; Al-Qenaei等人)。 (2014年)。我们认为高的胞质铁将催化PAA自氧化,导致细胞死亡。因为MM肿瘤细胞比非肿瘤细胞具有更高的不稳定铁池(LIP),所以我们假设PAA的抗癌作用取决于LIP。先前我们已经表明,Fpn1是唯一已知的哺乳动物铁输出蛋白,在MM细胞中的表达水平下调,导致LIP更高。接下来,我们试图确定MM肿瘤细胞中较高的Fpn1水平是否还能阻断PAA介导的细胞死亡。我们通过qRT-PCR在OCI-MY5细胞中过表达并确认了Fpn1的表达(补充图5B,OE-Fpn1)。我们注意到4
mM
PAA能够杀死用空载体(EV)转染的OCI-MY5,但不能杀死过度表达Fpn1(OE-Fpn1)细胞(图2B)。要成功杀死OE-Fpn1细胞,需要浓度提高五倍的PAA(20
mM)。由于Fpn1在OCI-MY5细胞中的过表达抑制了PAA的抗癌活性,因此我们接下来探讨了铁补充剂是否能够恢复对PAA的敏感性。铁预处理导致OCI-MY5
EV细胞活力迅速下降(图2C),并且在OE-Fpn1
OCI-MY5细胞中也获得了相同的效果(图2D)。与我们的假设一致,铁螯合剂DFO(图2C和D)取消了PAA降低用铁预处理的EV和OE-Fpn1
OCI-MY5细胞中细胞活力的能力。有趣的是,我们注意到仅用PAA处理的图2C中的EV
OCI-MY5细胞显示出比图2B中的那些细胞更强的敏感性。我们推测即使在两个实验中PAA温育为1小时,图2C中的细胞在培养中也比图2B中的细胞长24小时,以便在PAA处理之前与其他试剂一起温育。可能的解释是,与培养基一起长时间孵育可能会略微增加细胞铁的含量(Goto等,1983)。有证据支持添加DFO(图2C)或Fpn1过表达(图2D)可以挽救PAA诱导的MM细胞死亡,甚至对照组之间的细胞生存力也不同,因为DFO和Fpn1的过表达都会降低细胞铁。
3.5。凋亡诱导因子1在药理学抗坏血酸诱导的骨髓瘤细胞死亡中起关键作用
我们随后尝试确定PAA诱导线粒体介导的细胞凋亡的分子途径。我们的假设是线粒体通透性增加是死亡信号转导机制的触发因素。我们将注意力集中在凋亡诱导因子1(AIF1)上,因为AIF1以caspase依赖性和caspase依赖性方式诱导细胞死亡(Nikoletopoulou等,2013)。我们评估PAA诱导的MM细胞死亡是否至少部分取决于AIF1。我们生成具有AIF1敲低(shRNA-AIF1)或过表达(OE-AIF1)的OCI-MY5细胞。
AIF1-shRNA OCI-MY5细胞的活力(图4A,顶部柱状图)显着高于PAA处理后表达乱序序列的细胞(图4A,顶部柱状图),而OE-AIF1
OCI-MY5显示出显着性与空载体(EV)转染的PAA处理后的细胞相比,存活率(图4A,底部柱状图)低(图4A,底部柱状图)。人们普遍认为,AIF1必须先从线粒体上裂解并释放到细胞质,然后再转移到细胞核,以诱导导致细胞死亡的色谱分解(图4B)(Sevrioukova,2011)。因此,我们检查了PAA是否诱导了AIF1裂解。通过蛋白质印迹,用PAA处理的OCI-MY5细胞显示出AIF1裂解形式的增加(图4C)。Melphalan无法在OCI-MY5细胞中诱导AIF1裂解(图4C),可能是因为Melphalan是一种烷基化剂,并产生了许多带有DNA链间交联键(ICL)的DNA加合物(被认为是关键的细胞毒性损伤)(Spanswick等)
(2002年)。我们假设AIF1切割是由PAA与LIP反应形成ROS介导的。因此,我们在有或没有DFO的情况下孵育了OCI-MY5细胞,然后进行了PAA处理。
PAIF在DFO预处理的OCI-MY5细胞中孵育后,AIF1没有被切割,从而证实了LIP在此过程中的关键作用(图4D,白色箭头)。早期的研究表明,DFO可以耗尽LIP(Al-Qenaei等人,2014)。我们还测试了PAA和melphalan处理后磷酸化的γ-H2AX(DNA双向断裂的生物标志物)的水平,并确定PAA和高剂量的melphalan诱导了γ-H2AX。但是,较低剂量的苯丙氨酸马法兰治疗剂也能够诱导γ-H2AX(图4C)。这些数据支持了我们较早的体内数据(图1F),即较低剂量的PAA和美法仑的组合与单独美法仑的抑制作用相同或更大。通过免疫标记电子显微镜在有或没有PAA处理的情况下,在OCI-MY5细胞中检查AIF1的细胞定位。该染色表明,AIF1不仅位于线粒体中(如在未处理的细胞中所见)(图4E,左图),而且还位于PAA处理的OCI-MY5细胞中的细胞质和细胞核中(图4E,右图)。这些结果表明PAA通过与LIP反应并产生ROS诱导线粒体介导的凋亡,其中AIF1裂解对细胞死亡很重要。
4。讨论
高剂量维生素C已在多种癌症中进行了研究,并已显示出有争议的临床效果(Cameron和Pauling,1978; Cameron和Pauling,1976;
Creagan等,1979;
Moertel等,1985)。矛盾的临床结果可以至少部分地通过口服或静脉内施用维生素C的不同途径来解释。最近的报告表明,大剂量维生素C诱导癌细胞的细胞毒性需要一定的ROS浓度。
PAA生成抗坏血酸基和H2O2自由基会增加癌细胞中的ROS应激(Du等人,2012年)。这些研究包括临床前和临床研究均在实体瘤中进行,例如胶质母细胞瘤(Herst等,2012),胰腺癌(Du等,2015),卵巢癌(Ma等,2014),前列腺癌(Chen)。等人,2012,Pollard等人,2010),肝癌(Verrax和Calderon,2009),结肠癌(Pires等人,2016),间皮瘤(Ranzato等人,2011),乳腺癌(Yun等人)
(2015年),膀胱癌(Gilloteaux等人,2010年)和神经母细胞瘤(Deubzer等人,2010年)。缺乏报道表明,PAA可以用作治疗“液体”肿瘤的促氧化剂,其中肿瘤细胞被血液包围。即使在高剂量下,实体瘤和血液癌之间的这种环境差异也可能影响PAA对癌细胞死亡的功效,因为抗坏血酸生成的ROS在液体肿瘤中的渗透性比在实体瘤中容易得多。在这项研究中,我们报道了PAA在体外和体内杀死MM细胞方面均有效,在腹膜内给予每千克体重4
g的抗坏血酸后,其可产生20-40 mM的抗坏血酸和500
nM的抗坏血酸基团(Chen等)等,2008),在异种移植MM小鼠中。这些数据表明,由于肿瘤细胞与血浆之间的通讯,PAA相对于实体瘤可能显示出对血液癌的治疗优势。
我们已经显示,Fpn1在体外和体内调节MM细胞和LIP中的铁输出(Gu等,2015)。此外,铁蛋白还通过螯合氧化形式的游离铁以防止自由基的形成来调节LIP(Pantopoulos等人,2012)。我们的初步数据显示,与PAA处理后的野生型细胞相比,MMn细胞系OCI-MY5中Fpn1的过表达导致活力增加。我们假设表达Fpn1的MM细胞对PAA的敏感性较低,因为Fpn1降低了它们的胞质铁含量。为了测试对PAA的耐药性是否确实是由于胞质铁含量低引起的,我们通过将细胞与铁螯合剂去铁胺(DFO)预孵育来消除胞质铁。 DFO的膜渗透性差。但是,在包括表达Fpn1的细胞在内的多项研究中,它已被用于螯合细胞内铁(Delaby等,2005; Knutson等,2003)。与未经DFO预处理的细胞相比,经DFO预处理的ARP1 MM细胞(200μM,3 h)和PAA处理显示出更高的生存能力。这些结果强烈表明PAA杀死MM细胞的机制确实是铁依赖性的。此外,与正常浆细胞相比,Fpn1在CD138 +原代MM细胞中显着下调,而铁导入剂转铁蛋白受体1在CD138 + MM细胞中显着上调,进一步支持MM细胞的铁含量高于非肿瘤细胞。细胞。 PAA显示,几乎所有原发性MM患者和闷烧的MM,但不是MGUS患者,对MM细胞的杀伤作用均增加。这些结果表明,在SMM中使用PAA可能能够预防进展为症状性MM。
MM患者的高氧化应激和DNA损伤活性增加,而抗氧化酶水平降低(Mehdi等,2013)。几种自由基药物(例如As2O3和抗坏血酸)已用于治疗MM,其中As2O3产生ROS,而抗坏血酸充当抗氧化剂。在MM临床前和临床研究中,抗坏血酸用作辅助药物并显示有争议的结果(Harvey等,2009; Perrone等,2009; Held等,2013; Sharma等,2012; Nakano等。 (例如,2011年,高桥(Takahashi),2010年,Sharma等,2009年,Qazilbash等,2008年)。但是,这些测试均未使用抗坏血酸和静脉内给药的药理剂量。据报道,抗坏血酸通过其邻位二醇基团形成紧密但可逆的复合物,直接使硼替佐米的活性失活(Perrone等,2009; Harvey等,2009)。抗坏血酸与硼替佐米合用的剂量处于具有抗氧化作用的生理水平。由于上述可能的化学反应,我们未将PAA与硼替佐米合并使用。但是,我们的初步研究还表明,PAA可以克服MM细胞对硼替佐米的耐药性(数据未显示)。
我们的发现补充了已报道的研究,并使用临床样品进一步探讨了作用机理,其中我们观察到PAA杀死了高铁含量的肿瘤细胞,这表明铁可能是PAA细胞毒性的引发剂。此外,PAA与标准治疗药物(例如美法仑)的组合可能会大大减少所需的美法仑的剂量。这是有益的,因为高剂量的美法仑不仅对肿瘤细胞有毒性,而且对正常组织如肠道造血干细胞和上皮细胞也有剧毒(Shaw等人,2014;
Bayraktar等人,2013)。高剂量马法兰的功效显然是剂量依赖性的。与单独使用相同剂量的美法仑相比,降低剂量的美法仑与PAA的联合治疗可显着延长无进展生存期。这些数据还表明,由于PAA对低铁含量的正常细胞缺乏毒性,因此可以通过将PAA与较低剂量的马法兰结合来改善高剂量马法兰的骨髓抑制作用。对于将来的临床研究,重要的是要考虑肾功能不全可能是MM患者使用PAA的禁忌证。肾功能不全是MM的常见并发症(50%),高达5%需要透析(Yadav等,2016a,Yadav等,2016b)。临床毒性可能来自草酸,这是抗坏血酸代谢的最终产物。因此,如果肌酐清除率<30
mL / min,则不应服用高剂量的抗坏血酸。
Multiple Myeloma Tumor Cells are Selectively Killed by Pharmacologically-dosed Ascorbic Acid
EBioMedicine. 2017 Apr; 18: 41–49. Abstract High-dose chemotherapies to treat multiple myeloma (MM) can be life-threatening due to toxicities to normal cells and there is a need to target only tumor cells and/or lower standard drug dosage without losing efficacy. We show that pharmacologically-dosed ascorbic acid (PAA), in the presence of iron, leads to the formation of highly reactive oxygen species (ROS) resulting in cell death. PAA selectively kills CD138+ MM tumor cells derived from MM and smoldering MM (SMM) but not from monoclonal gammopathy undetermined significance (MGUS) patients. PAA alone or in combination with melphalan inhibits tumor formation in MM xenograft mice. This study shows PAA efficacy on primary cancer cells and cell lines in vitro and in vivo. Keywords: Multiple myeloma, Pharmacologically-dosed ascorbic acid, Apoptosis-inducing factor 1 Abbreviation: MM, multiple myeloma; ROS, reactive oxygen species; SMM, smoldering MM; IMIDs, immunomodulatory drugs; ASCT, autologous stem cell transplantation; CRs, complete remissions; PAA, pharmacologically dosed ascorbic acid; LIP, labile iron pool; GEP, gene expression profiling; BM, bone marrow; IVIS, in vivo imaging system; WT, wild-type; DFO, deferoxamine; AIF1, apoptosis inducible factor 1 Go to: 1. Introduction Multiple myeloma (MM) is a plasma cell neoplasm. Four active classes of drugs including glucocorticoids, DNA alkylators (melphalan), proteasome inhibitors (bortezomib and carfilzomib) and immunomodulatory agents (thalidomide, lenalidomide, and pomalidomide), combined with or without autologous stem cell transplantation (ASCT) have led to complete remissions (CRs) in the large majority of newly diagnosed patients with MM (Alexanian et al., 2014, Fu et al., 2013, Terpos et al., 2014, Wang et al., 2014, Sonneveld et al., 2013, Gay et al., 2013, Liu et al., 2013, Bergsagel, 2014). These treatments have greatly improved patient progression-free and overall survival. However, there are at least three major problems limiting the administration of these agents: 1. All these drugs target both tumor and non-tumor cells; 2. Increased hematologic toxicity has been identifined by combining alkylators with either immunomodulatory drugs (IMIDs) (Bergsagel, 2014); and 3. High doses of the DNA alkalating agent, such as melphalan, have strong cytotoxicity on gut epithelial cells and hematopoietic stem cells (Shaw et al., 2014). One way to deal with non-selective toxicity of high dose melphalan is to combine it with another agent which very specifically targets tumor cells and therefore decreasing melphalan dosing without loss of efficacy.
In the 1970s, Cameron and Pauling reported that high doses of vitamin C
increased survival of patients with cancer (Cameron and Pauling, 1976, Cameron
and Pauling, 1978). Recently, reports have shown that pharmacologically dosed
ascorbic acid (PAA) 50–100 g (Chen et al., 2008, Padayatty et al., 2004, Hoffer
et al., 2008, Padayatty et al., 2006, Welsh et al., 2013), administered
intravenously, has potent anti-cancer activity and its role as anti-cancer
therapy is being studied at the University of Iowa and in other centers (Du et
al., 2012, Ma et al., 2014). In the presence of catalytic metal ions like iron,
PAA administered intravenously exerts pro-oxidant effects leading to the
formation of highly reactive oxygen species (ROS), resulting in cell death (Yun
et al., 2015, Ma et al., 2014, Du et al., 2012, Chen et al., 2007, Chen et al.,
2005). In a previous study, we have reported that the labile iron pool (LIP) is
significantly elevated in MM cells (Gu et al., 2015), suggesting that PAA
treatment should target MM cells quite selectively. The higher LIP is the direct
result of the low expression of the only known mammalian cellular iron exporter,
Ferroportin 1 (Fpn1), in MM as demonstrated by our group (Gu et al., 2015).
These findings led us to the hypothesis that PAA might specifically target MM
cells with high iron content and may also act synergistically in combination
with commonly used MM therapies.
RESULTS
3.3. The Therapeutic Effect of Pharmacological Ascorbic Acid Depends on
Cellular Iron and Reactive Oxygen Species
We subsequently asked whether PAA was selectively killing MM tumor cells by
generating ROS, we treated OCI-MY5 MM wild-type (WT) cells with N-acetyl
cysteine (NAC) or catalase. Both catalase and NAC protect cells from oxidative
damage. OCI-MY5 cells pretreated with NAC and catalase became resistant to PAA
even at high doses (Fig. 2A). Importantly, adding deferoxamine (DFO), an iron
chelator, to OCI-MY5 cells before PAA treatment was also sufficient to prevent
PAA-induced cellular death (Fig. 2A) but bathocuproinedisulfonic acid disodium
salt (BCS), a selective copper chelator, was not able to block MM cell death
(Supplementary Fig. 5A) suggesting that iron is essential for PAA to achieve its
anti-cancer activity. DFO is a poorly membrane permeable iron chelator, however,
it has been used to chelate intracellular iron in multiple studies including
those ones that describe cells expressing Fpn1(Asano et al., 2011, Paradkar et
al., 2008, Al-Qenaei et al., 2014). We reasoned that high cytosolic iron would
catalyze PAA auto-oxidation leading to cell death. Because MM tumor cells have a
higher labile iron pool (LIP) than non-tumor cells, we hypothesized that PAA's
anti-cancer effect is dependent on LIP. We have previously shown that Fpn1, the
only known mammalian iron exporter, is down-regulated in MM cells at the
expression levels leading to higher LIP. We next sought to determine if higher
Fpn1 levels in MM tumor cells could also block cell death mediated by PAA. We
overexpressed and confirmed Fpn1 expression by qRT-PCR in OCI-MY5 cells
(Supplementary Fig. 5B, OE-Fpn1). We noticed that 4 mM PAA was able to kill
OCI-MY5 transfected with empty vector (EV) but not to overexpressing Fpn1
(OE-Fpn1) cells (Fig. 2B). Five-fold greater concentration of PAA (20 mM) was
required to successfully kill OE-Fpn1 cells. Since the overexpression of Fpn1 in
OCI-MY5 cells inhibits PAA anti-cancer activity, we next explored whether iron
supplementation was able to restore sensitivity to PAA. Iron pre-treatment
caused a rapid decrease in cell viability of OCI-MY5 EV (Fig. 2C) and the same
effect was obtained in OE-Fpn1 OCI-MY5 cells (Fig. 2D). Consistent with our
hypothesis, DFO, an iron chelator (Fig. 2C & D), abolished the ability of PAA to
reduce cells viability in both EV and OE-Fpn1 OCI-MY5 cells pre-treated with
iron. Interestingly, we noticed that EV OCI-MY5 cells in Fig. 2C treated with
PAA alone showed a stronger sensitivity than those cells in Fig. 2B. We
speculated that even if the PAA incubation is 1 h in both experiments the cells
in Fig. 2C were kept in culture almost 24 h longer than cells in Fig. 2B for
incubation with other reagents before PAA treatment. A possible explanation is
that the longer incubation with culture media may slightly increase cellular
iron (Goto et al., 1983). This was supported by the evidence that either
addition of DFO (Fig. 2C) or overexpression of Fpn1 (Fig. 2D) rescued
PAA-induced MM cell death even cell viability was different between control
groups, because both DFO and overexpression of Fpn1 decrease cellular iron.
3.5. Apoptosis-inducing Factor 1 Plays a Critical Role in Pharmacological
Ascorbic Acid-induced Myeloma Cell Death
We subsequently tried to determine the molecular pathway by which PAA induced
mitochondria-mediated apoptosis. Our hypothesis was that increased mitochondrial
permeabilization was the trigger for the death signal transduction machinery. We
focused our attention on apoptosis-inducing factor 1 (AIF1), because AIF1
induces cell death in caspase-dependent and caspase-independent manners
(Nikoletopoulou et al., 2013). We evaluate if PAA induced MM cell death depends
on AIF1 at least partially. We generated OCI-MY5 cells with AIF1 knockdown
(shRNA-AIF1) or overexpression (OE-AIF1). The viability of AIF1-shRNA OCI-MY5
cells (Fig. 4A, top bar graph) was significantly higher than those cells
expressing scrambled sequence after PAA treatment (Fig. 4A, top bar graph),
while OE-AIF1 OCI-MY5 showed significantly less viability (Fig. 4A, bottom bar
graph) than cells transfected with empty vector (EV) when treated with PAA (Fig.
4A, bottom bar graph). It is widely accepted that AIF1 must be cleaved and
released from the mitochondria to the cytoplasm and then translocate to the
nucleus to induce chromatolysis leading to cell death (Fig. 4B) (Sevrioukova,
2011). We thus examined if PAA induced AIF1 cleavage. OCI-MY5 cells treated with
PAA showed an increase in the AIF1 cleaved form by western blotting (Fig. 4C).
Melphalan was not able to induce AIF1 cleavage in OCI-MY5 cells (Fig. 4C)
probably because melphalan is an alkylating agent and produces a number of DNA
adducts with DNA interstrand crosslinks (ICLs) considered to be the critical
cytotoxic lesion (Spanswick et al., 2002) in an AIF1 independent mechanism. We
hypothesized that the AIF1 cleavage was mediated by PAA reacting with LIP to
form ROS. Thus, we incubated OCI-MY5 cells with or without DFO followed by PAA
treatment. AIF1 was not cleaved after PAA incubation in OCI-MY5 cells pretreated
with DFO confirming the crucial role of LIP in this process (Fig. 4D, white
arrow). Earlier studies have shown that DFO is able to deplete LIP (Al-Qenaei et
al., 2014). We also tested the level of phosphorylated γ-H2AX, a biomarker for
DNA double-stand breaks, after PAA and melphalan treatment, and determined that
PAA and high dose of melphalan induced γ-H2AX. However, a lower dose of
melphalan with PAA was also able to induce γ-H2AX (Fig. 4C). These data support
our earlier in vivo data (Fig. 1F) that combination of PAA and melphalan at
lower dose inhibits tumor formation at the same level or greater than melphalan
alone. Cellular localization of AIF1 was examined by immunolabeling electron
microscope with and without PAA treatment in OCI-MY5 cells. This staining
revealed that AIF1 localizes not only in the mitochondria, as seen in untreated
cells (Fig. 4E, left panel), but also in cytoplasm and nuclei in PAA-treated
OCI-MY5 cells (Fig. 4E, right panel). These results indicate that PAA by
reacting with LIP and generating ROS induces mitochondria-mediated apoptosis in
which AIF1 cleavage is important for cell death.
4. Discussion
High-dose vitamin C has been studied in multiple cancers and has shown
controversial clinical effects (Cameron and Pauling, 1978, Cameron and Pauling,
1976, Creagan et al., 1979, Moertel et al., 1985). The contradictory clinical
results can be at least partially explained by different routes of vitamin C
administration applied, i.e., either orally or intravenously. Recent reports
indicate that a certain ROS concentration is required for high-dose vitamin C to
induce cytotoxicity in cancer cells. The generation of ascorbyl- and H2O2
radicals by PAA increases ROS stress in cancer cells (Du et al., 2012). These
studies including preclinical and clinical were performed in solid tumors, such
as glioblastoma (Herst et al., 2012), pancreatic cancer (Du et al., 2015),
ovarian cancer (Ma et al., 2014), prostate cancer (Chen et al., 2012, Pollard et
al., 2010), hepatoma (Verrax and Calderon, 2009), colon cancer (Pires et al.,
2016), mesothelioma (Ranzato et al., 2011), breast cancer (Yun et al., 2015),
bladder cancer (Gilloteaux et al., 2010), and neuroblastoma (Deubzer et al.,
2010). Reports are lacking to show that PAA can be used as a pro-oxidant drug in
the treatment of “liquid” tumors, where tumor cells are surrounded by blood.
This environmental difference between solid tumor and blood cancer has the
potential to influence the PAA efficacy on cancer cell death even when given at
high doses, because ascorbic acid generated ROS are much easier permeabilized in
liquid tumor than in solid tumor. In this study, we report that PAA is
efficacious in killing MM cells in vitro and in vivo models, which generated
levels of 20–40 mM ascorbate and 500 nM ascorbyl radicals after intraperitoneal
administration of 4 g ascorbate per kilogram of body weight (Chen et al., 2008),
in xenograft MM mice. These data suggest that PAA may show a therapeutic
advantage to blood cancers vs solid tumors because of the communication between
tumor cells and blood plasma.
We have shown that Fpn1 regulates iron export in MM cells and LIP in vitro and in vivo (Gu et al., 2015). In addition, ferritin also regulates LIP by sequestering free iron in an oxidized form to prevent formation of free radicals (Pantopoulos et al., 2012). Our preliminary data show that overexpression of Fpn1 in MM cell line OCI-MY5 results in increased viability compared to wild type cells after PAA treatment. We hypothesize that Fpn1 expressing MM cells are less sensitive to PAA because their cytosolic iron content is reduced by Fpn1. To test if resistance to PAA is indeed due to low cytosolic iron content, we depleted cytosolic iron by pre-incubating cells with an iron chelator, deferoxamine (DFO). DFO is poorly membrane permeable. However, it has been used to chelate intracellular iron in multiple studies including cells expressing Fpn1 (Delaby et al., 2005, Knutson et al., 2003). ARP1 MM cells pre-treated with DFO (200 μM, 3 h) followed by PAA treatment showed a higher viability than cells not pre-treated with DFO. These results strongly suggest that the mechanism of PAA killing of MM cells is indeed iron-dependent. In addition, Fpn1 is significantly down-regulated in CD138+ primary MM cells, while the iron importer, transferrin receptor 1, is significantly upregulated in CD138+ MM cells compared to normal plasma cells, further supporting that MM cells have higher iron content than non-tumor cells. PAA showed increased killing of MM cells derived from almost all primary MM patients and smoldering MM, but not from MGUS patients. These results suggest that PAA administration in SMM may be able to prevent progression to symtomatic MM.
Our findings complement reported studies and further address the mechanism of action using clinical samples in which we observed that PAA killed tumor cells with high iron content, suggesting that iron might be the initiator of PAA cytotoxicity. In addition, combination of PAA with standard therapeutic drugs, such as melphalan, may significantly reduce the dose of melphalan needed. This is beneficial because high doses of melphalan are very toxic not only to tumor cells but also to normal tissues, such as hematopoietic stem cell and epithelial cells in the gut (Shaw et al., 2014, Bayraktar et al., 2013). The efficacy of high-dose melphalan by itself is clearly dose-dependent. Combined treatment of reduced dose melphalan with PAA achieved a significantly longer progression-free survival than the same dose of melphalan alone. These data also suggest that the bone marrow suppression induced by high-dose melphalan can be ameliorated by the combination of PAA with lower dose of melphalan because of the lack of toxicity of PAA on normal cells with low iron content. It is important to consider for future clinical studies that renal insufficiency could be a contraindication for usage of PAA in MM patients. Renal impairment is a common complication of MM (50%) and up to 5% require dialysis (Yadav et al., 2016a, Yadav et al., 2016b). The clinical toxicity is probably from oxalate, an end-product of ascorbate metabolism. Therefore, if creatinine clearance is < 30 mL/min, high dose ascorbic acid should be not administrated.
Multiple Myeloma Tumor Cells are Selectively Killed by
Pharmacologically-dosed Ascorbic Acid
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5405162/
维生素C是一种激酶抑制剂:脱氢抗坏血酸抑制IκBα激酶β,因此NF-kB的激活
Vitamin C Is a Kinase Inhibitor: Dehydroascorbic Acid Inhibits IκBα Kinase β
Department of Clinical Laboratories,1 Program in Molecular Pharmacology and Chemistry, Memorial Sloan-Kettering Cancer Center,2 Structural Biology Program, Department of Physiology and Biophysics, Mount Sinai School of Medicine, New York, New York3
活性氧(ROS)是细胞信号转导通路中的关键中间体,其功能可能会被抗氧化剂抵消。抗坏血酸(AA)充当抗氧化剂,提供两个电子并被氧化成脱氢抗坏血酸(DHA)。我们发现DHA在体外直接抑制IκBα激酶β(IKKβ)和IKKα的酶活性,而AA则没有这种作用。当在细胞中加载AA并通过表达组成型活性IKKβ的细胞中的氧化应激诱导生成DHA时,NF-κB激活受到抑制。我们的结果确定了维生素C在信号转导中的双重分子作用,并提供了维生素C的氧化还原状态与NF-κB信号事件之间的直接联系。
AA淬灭参与NF-κB活化的ROS中间体,并被氧化成DHA,直接抑制IKKβ和IKKα的酶活性。这些发现定义了维生素C在信号传导中的功能,除了作为抗氧化剂外,还从机理上阐明了维生素C如何下调NF-κB信号传导。
我们调查了ROS的细胞内生成器,例如二甲氧萘并萘醌(DMNQ),是否还会抑制预加载了2.5 mM维生素C的细胞中的IKKβ(SS /
EE)依赖性荧光素酶活性。DMNQ的浓度对荧光素酶活性或发现用IKKβ(SS / EE)转染的细胞的生存力为5μM。装有2.5
mM维生素C并与5μMDMNQ孵育的细胞显示萤光素酶活性显着降低(P = 0.003,n =
3,单尾t检验)(图(图6B).6B)。这些结果表明,AA向DHA的细胞内转化抑制了IKKβ(SS /
EE)的激酶活性,表明DHA在体内起激酶抑制剂的作用。我们认为,加载维生素C并置于氧化条件下的细胞中,AA可以抑制ROS并转化为DHA。生成的DHA可以通过促进性葡萄糖转运蛋白离开细胞,或作为直接激酶抑制剂发挥作用,如此处所示,用于IKKα和IKKβ(图(图7).7)。这些过程通过抗氧化剂维生素C将氧化应激与激酶抑制联系起来。

图。 7。
维生素C调节信号响应的示意图。维生素C作为DHA通过葡萄糖转运蛋白进入细胞,并迅速还原为AA。
ROS通过激活IKKβ诱导NF-κB信号传导应答,而AA猝灭ROS,抑制IKKβ的激活。在这些过程中,AA氧化成DHA,而DHA抑制IKKβ。
Vitamin C Is a Kinase Inhibitor: Dehydroascorbic Acid Inhibits IκBα Kinase β
Reactive oxygen species (ROS) are key intermediates in cellular signal
transduction pathways whose function may be counterbalanced by antioxidants.
Acting as an antioxidant, ascorbic acid (AA) donates two electrons and becomes
oxidized to dehydroascorbic acid (DHA). We discovered that DHA directly inhibits
IκBα kinase β (IKKβ) and IKKα enzymatic activity in vitro, whereas AA did not
have this effect. When cells were loaded with AA and induced to generate DHA by
oxidative stress in cells expressing a constitutive active IKKβ, NF-κB
activation was inhibited. Our results identify a dual molecular action of
vitamin C in signal transduction and provide a direct linkage between the redox
state of vitamin C and NF-κB signaling events. AA quenches ROS intermediates
involved in the activation of NF-κB and is oxidized to DHA, which directly
inhibits IKKβ and IKKα enzymatic activity. These findings define a function for
vitamin C in signal transduction other than as an antioxidant and
mechanistically illuminate how vitamin C down-modulates NF-κB signaling.
We investigated whether an intracellular generator of ROS, such as
dimethoxinaphthoquinine (DMNQ), would also inhibit IKKβ(SS/EE)-dependent
luciferase activity in cells preloaded with 2.5 mM vitamin C. The concentration
of DMNQ that had no effect on luciferase activity or viability in cells
transfected with IKKβ(SS/EE) was found to be 5 μM. Cells loaded with 2.5 mM
vitamin C and incubated with 5 μM DMNQ showed significantly reduced luciferase
activity (P = 0.003, n = 3, one-tailed t test) (Fig. (Fig.6B).6B). These
results indicate that intracellular conversion of AA to DHA inhibits kinase
activity of IKKβ(SS/EE), suggesting that DHA functions as a kinase inhibitor in
vivo. We propose that in cells loaded with vitamin C and placed under oxidative
conditions, AA quenches ROS and transforms into DHA. The DHA generated can leave
the cell via facilitative glucose transporters or function as a direct kinase
inhibitor as shown here for IKKα and IKKβ (Fig. (Fig.7).7). These processes
link oxidative stress to kinase inhibition via the antioxidant, vitamin C.
Vitamin C Is a Kinase Inhibitor: Dehydroascorbic Acid Inhibits IκBα Kinase β
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC444845/
Vitamin C Is a Kinase Inhibitor: Dehydroascorbic Acid Inhibits IκBα Kinase β
| Molecular and Cellular Biology
https://mcb.asm.org/content/24/15/6645
脱氢抗坏血酸的新型生物学作用:抑制Na +依赖性抗坏血酸的转运
A novel biological role of dehydroascorbic acid: Inhibition of Na+-dependent transport of ascorbic acid
Dipartimento di Scienze Biomolecolari, Università degli Studi di Urbino
“Carlo Bo”, Urbino 61029, Italy
使用U937细胞克隆,以相同的速率吸收低微摩尔浓度的抗坏血酸(AA)和脱氢抗坏血酸(DHA),用于研究介导两种形式维生素的细胞吸收的转运系统之间可能的相互作用。用不同的实验方法获得的结果表明,DHA有效且可逆地抑制了Na
+
-AA共转运蛋白对AA的吸收。因此,在固定量的AA存在下,细胞外DHA浓度的逐渐升高会导致维生素C净积累量的初始下降,最终,在较高水平下,仅基于DHA摄取会导致维生素的积累通过己糖转运蛋白。在其他各种细胞类型中也检测到了DHA依赖的AA吸收抑制作用。综上所述,我们的结果提供了由与炎症部位产生的DHA浓度相适应的DHA浓度介导的新型生物学效应的证据。
.jpg)
A novel biological role of dehydroascorbic acid: Inhibition of Na+-dependent
transport of ascorbic acid
A U937 cell clone, in which low micromolar concentrations of ascorbic acid (AA)
and dehydroascorbic acid (DHA) are taken up at identical rates, was used to
investigate possible interactions between transport systems mediating cellular
uptake of the two forms of the vitamin. Results obtained with different
experimental approaches showed that DHA potently and reversibly inhibits AA
uptake through Na+-AA cotransporters. Hence, a progressive increase in
extracellular DHA concentrations in the presence of a fixed amount of AA caused
an initial decrease in the net amount of vitamin C accumulated, and eventually,
at higher levels, it caused an accumulation of the vitamin solely based on DHA
uptake through hexose transporters. DHA-dependent inhibition of AA uptake was
also detected in various other cell types. Taken together, our results provide
evidence of a novel biological effect mediated by concentrations of DHA
compatible with those produced at inflammatory sites.
A novel biological role of dehydroascorbic acid: Inhibition of Na+-dependent
transport of ascorbic acid - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S1043661814000462
黄酮类化合物阻断脱氢抗坏血酸和抗坏血酸的摄取,抑制细胞内AA和DHA在积累U937和Jurkat细胞中的积累
Intracellular Accumulation of Ascorbic Acid Is Inhibited by Flavonoids via Blocking of Dehydroascorbic Acid and Ascorbic Acid Uptakes in HL-60, U937 and Jurkat Cells
马克·莱文(James B.
营养学杂志,第130卷,第5期,2000年5月,第1297页
抽象
在HL-60,U937和Jurkat细胞中,细胞内抗坏血酸的积累是通过摄取脱氢抗坏血酸(抗坏血酸的氧化代谢产物)和抗坏血酸(维生素C)而发生的。脱氢抗坏血酸和抗坏血酸分别通过钠依赖性葡萄糖转运蛋白(GLUT
1和GLUT 3)和钠依赖性抗坏血酸转运蛋白转运到细胞中。
类黄酮通过阻断转化细胞中的脱氢抗坏血酸和抗坏血酸的摄取来抑制细胞内抗坏血酸的积累。当类黄酮浓度为10-70μmol/ L时,细胞中约50%的脱氢抗坏血酸摄取被抑制。在Jurkat细胞中,两种有效的类黄酮(杨梅素和槲皮素)竞争性地抑制脱氢抗坏血酸的摄取,Ki值分别约为14和15μmol/ L。因为GLUT 1和GLUT 3转运脱氢抗坏血酸,所以使用过表达大鼠GLUT 1或人GLUT 3的中国仓鼠卵巢细胞研究了黄酮类化合物对脱氢抗坏血酸摄取的抑制作用。杨梅素分别抑制22和18μmol/ L的浓度在过表达GLUT 1和GLUT 3的细胞中脱氢抗坏血酸的摄取。杨梅素还抑制抗坏血酸的摄取。在Jurkat细胞中,Ki = 14μmol/ L时,抑制作用是非竞争性的。
这些数据表明类黄酮抑制抗坏血酸和脱氢抗坏血酸的摄取,但是通过不同的机制抑制。这些数据可能有助于对类黄酮对人体细胞中抗坏血酸细胞内积累的生物学作用的新认识。
Intracellular Accumulation of Ascorbic Acid Is Inhibited by Flavonoids via
Blocking of Dehydroascorbic Acid and Ascorbic Acid Uptakes in HL-60, U937 and
Jurkat Cells
Jae B. Park, Mark Levine
The Journal of Nutrition, Volume 130, Issue 5, May 2000, Pages 1297–
ABSTRACT
In HL-60, U937 and Jurkat cells, the intracellular accumulation of ascorbic acid
occurred via uptakes of both dehydroascorbic acid (an oxidized metabolite of
ascorbic acid) and ascorbic acid (vitamin C). Dehydroascorbic acid and ascorbic
acid were transported into cells by sodium-independent glucose transporters
(GLUT 1 and GLUT 3) and sodium-dependent ascorbic acid transporters,
respectively.
Flavonoids inhibited the intracellular accumulation of ascorbic acid by blocking dehydroascorbic acid and ascorbic acid uptakes in the transformed cells.
At flavonoid concentrations of 10–70 μmol/L, ∼50% of dehydroascorbic acid uptake was inhibited in the cells. In Jurkat cells, two potent flavonoids (myricetin and quercetin) competitively inhibited dehydroascorbic acid uptake, and Ki values were ∼14 and 15 μmol/L, respectively. Because GLUT 1 and GLUT 3 transport dehydroascorbic acid, the inhibition of dehydroascorbic acid uptake by flavonoids was investigated by using Chinese hamster ovary cells overexpressing rat GLUT 1 or human GLUT 3. Myricetin at concentrations of 22 and 18 μmol/L, respectively, inhibited half of dehydroascorbic acid uptake in the cells overexpressing GLUT 1 and GLUT 3. Myricetin also inhibited ascorbic acid uptake; inhibition was noncompetitive with Ki = 14 μmol/L in Jurkat cells.
These data indicate that flavonoids inhibit both ascorbic acid and dehydroascorbic acid uptake but do so by different mechanisms. These data may contribute to new understanding of the biological effect of flavonoids on the intracellular accumulation of ascorbic acid in human cells.
Reduction of dehydroascorbic acid by homocysteine
Author links open overlay panelJae BPark
Phytonutrients Laboratory, Bldg. 307, Rm. 313, BHNRC, ARS, USDA, Beltsville, MD
20705, USA
Abstract
To determine the reductive process of extracellular dehydroascorbic acid (DHA),
molecules (homocysteine, homocysteine thiolactone, methionine, cysteine, and
homoserine) were tested to identify those with the potential to reduce DHA to
ascorbic acid (AA). Homocysteine (Hcy) was the most potent of the molecules
tested. The efficacy of Hcy was compared with that of other molecules able to
reduce DHA (reduced glutathione (GSH) and cysteine (Cy)). Although all three
molecules were able to reduce DHA, GSH and Cy were not to reduce DHA to AA at
concentrations lower than 100 μmol/l, and only less than 5% DHA was reduced to
AA at concentrations of 200–300 μmol/l. In contrast, Hcy reduced DHA to AA
stoichiometrically at concentrations as low as 10 μmol/l. In Jurkat and U937
cells, the increasing concentrations of extracellular Hcy suppressed
intracellular dehydroascorbic acid uptake, indicating that extracellular
reduction of DHA by Hcy leads to decreasing extracellular DHA available for its
intracellular uptake. Simultaneous oxidation and reduction of Hcy and DHA were
accelerated extracellularly in the presence of quercetin, an inhibitor of DHA
uptake, suggesting that extracellular ascorbic acid concentration increased via
blocking DHA uptake by quercetin and reducing extracellular DHA by Hcy. The
effect of homocysteine on DHA reduction and uptake was confirmed with human
umbilical vein endothelial cells. The oxidation of Hcy also prevented the
decrease in DNA synthesis in human umbilical vein endothelial cells, which would
occur following exposure to Hcy.
Reduction of dehydroascorbic acid by homocysteine - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S0304416500001860
AMP-activated protein kinase inhibits NF-κB signaling and
...
https://paperity.org/p/35744757/amp-activated-protein-kinase-inhibits-nf-kb-signaling...
Cacicedo et al. [45] observed that AMPK activation inhibits NF-B transactivation
induced by TNF- and fatty acid palmitate in endothelial cells. In addition, Fig.
1 Schematic illustration of the functional connections of AMPK linked to the
inhibition of NF-B signaling and suppression of inflammation.
MP-activated protein kinase inhibits NF-κB signaling and inflammation: impact on
healthspan and lifespan
Journal of Molecular Medicine, Jul 2011 Antero Salminen Juha M. T. Hyttinen Kai Kaarniranta
Adenosine monophosphate-activated protein kinase (AMPK) is a crucial regulator of energy metabolic homeostasis and thus a major survival factor in a variety of metabolic stresses and also in the aging process.
Metabolic syndrome is associated with a low-grade, chronic inflammation, primarily in adipose tissue. A low-level of inflammation is also present in the aging process. There are emerging results indicating that AMPK signaling can inhibit the inflammatory responses induced by the nuclear factor-κB (NF-κB) system. The NF-κB subunits are not direct phosphorylation targets of AMPK, but the inhibition of NF-κB signaling is mediated by several downstream targets of AMPK, e.g., SIRT1, PGC-1α, p53, and Forkhead box O (FoxO) factors.
AMPK signaling seems to enhance energy metabolism while it can repress inflammatory responses linked to chronic stress, e.g., in nutritional overload and during the aging process. AMPK can inhibit endoplasmic reticulum and oxidative stresses which are involved in metabolic disorders and the aging process. Interestingly, many target proteins of AMPK are so-called longevity factors, e.g., SIRT1, p53, and FoxOs, which not only can increase the stress resistance and extend the lifespan of many organisms but also inhibit the inflammatory responses. The activation capacity of AMPK declines in metabolic stress and with aging which could augment the metabolic diseases and accelerate the aging process. We will review the AMPK pathways involved in the inhibition of NF-κB signaling and suppression of inflammation. We also emphasize that the capacity of AMPK to repress inflammatory responses can have a significant impact on both healthspan and lifespan.
AMP-activated protein kinase inhibits NF-κB signaling and inflammation:
impact on healthspan and lifespan (pdf) | Paperity
https://paperity.org/p/35744757/amp-activated-protein-kinase-inhibits-nf-kb-signaling-and-inflammation-impact-on
Inhibition of NF-κB Signaling as a Strategy in Disease Therapy
https://link.springer.com/chapter/10.1007/82_2010_105
Nov 27, 2010 · Gilmore T.D., Garbati M.R. (2010) Inhibition of NF-κB Signaling
as a Strategy in Disease Therapy. In: Karin M. (eds) NF-kB in Health and
Disease. Current Topics in …
IκB kinase β (IKKβ/IKK2/IKBKB)—A key molecule in signaling ...
https://www.academia.edu/8766751/IκB_kinase_β_IKKβ_IKK2_IKBKB_A_key_molecule_in...
The kinase activity of IKKb targets two adjacent serine residues of IkB leading
to ubiquitination and proteasomal degradation of the inhibitor, followed by
release and activation of NF-kB. Many signaling pathways that activate NF-kB
converge at the level of IKKb.
J Immunol. 2000 Dec 15;165(12):7180-8.
Vitamin C inhibits NF-kappa B activation by TNF via the
activation of p38 mitogen-activated protein kinase.
Bowie AG1, O'Neill LA.
Author information
1
Department of Biochemistry, Trinity College, Dublin, Ireland. agbowie@tcd.ie
Abstract
The transcription factor NF-kappaB is a central mediator of altered gene
expression during inflammation, and is implicated in a number of pathologies,
including cancer, atherosclerosis, and viral infection. We report in this study
that vitamin C inhibits the activation of NF-kappaB by multiple stimuli,
including IL-1 and TNF in the endothelial cell line ECV304 and in primary
HUVECs. The induction of a NF-kappaB-dependent gene, IL-8, by TNF was also
inhibited. The effect requires millimolar concentrations of vitamin C, which
occur intracellularly in vivo, particularly during inflammation. Vitamin C was
not toxic to cells, did not inhibit another inducible transcription factor,
STAT1, and had no effect on the DNA binding of NF-kappaB. Inhibition by vitamin
C was not simply an antioxidant effect, because redox-insensitive pathways to
NF-kappaB were also blocked. Vitamin C was shown to block IL-1- and TNF-mediated
degradation and phosphorylation of I-kappaBalpha (inhibitory protein that
dissociates from NF-kappaB), due to inhibition of I-kappaB kinase (IKK)
activation. Inhibition of TNF-driven IKK activation was mediated by p38
mitogen-activated protein kinase, because treatment of cells with vitamin C
led to a rapid and sustained activation of p38, and the specific p38 inhibitor
SB203580 reversed the inhibitory effect of vitamin C on IKK activity,
I-kappaBalpha phosphorylation, and NF-kappaB activation. The results identify
p38 as an intracellular target for high dose vitamin C.
Vitamin C inhibits NF-kappa B activation by TNF via the activation of p38
mitogen-activated protein kinase. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/11120850
The pathways to tumor suppression via route p38
Besides its well-known functions in inflammation and other stresses, the p38
mitogen-activated protein kinase pathway also negatively regulates cell
proliferation and tumorigenesis. Inactivation of the p38 pathway enhances
cellular transformation and renders mice prone to tumor development with
concurrent disruption of the induction of senescence. Conversely, persistent
activation of p38 inhibits tumorigenesis. Mechanistic insights into this
additional p38 function are starting to emerge. For example, p38 has been shown
to have a crucial role in oncogene-induced senescence, replicative senescence,
DNA-damage responses and contact-inhibition. In addition, the role of the p38
pathway in proliferative control and tumor suppression is mediated by its impact
on several cell-cycle regulators. These findings reveal a tumor-suppressing
function of the p38 pathway, and indicate that components of the p38 pathway are
potential targets for novel cancer therapies.
Key Laboratory of Ministry of Education for Cell Biology and Tumor Cell
Engineering, School of Life Sciences, Xiamen University, Xiamen 361005, China
Department of Molecular Biology, The Scripps Research Institute, 10550 North
Torrey Pines Road, La Jolla, CA 92037, USA
The pathways to tumor suppression via route p38: Trends in Biochemical Sciences
https://www.cell.com/trends/biochemical-sciences/fulltext/S0968-0004(07)00149-1
A Radical Role for p38 MAPK in Tumor Initiation
Norman J. Kennedy
Cristina Cellurale
Roger J. Davis
Program in Molecular Medicine, University of Massachusetts Medical School,
Worcester, MA 01605, USA
It is established that p38 MAPK can negatively regulate tumorigenesis, but the
mechanism is incompletely understood. A new study in this issue of Cancer Cell
shows that p38 MAP kinase plays a selective role in tumor initiation mediated by
oxidative stress.
Cells sense changes in their environment by activating signal transduction
pathways that direct biochemical programs to mediate proliferation,
differentiation, and survival. The mitogen-activated protein kinase (MAPK)
family represents an important group of signaling proteins that can regulate
these fundamental cellular processes.
The extracellular signal-regulated kinase (ERK) MAPK pathway primarily
directs a program of proliferation and survival, while the cJun NH2-terminal
kinase (JNK) pathway can promote either proliferation or apoptosis (Kennedy and
Davis, 2003). Conversely, the p38 MAPK pathway is activated upon cellular stress
and often engages pathways that can block proliferation or promote apoptosis
(Bulavin and Fornace, 2004).
The importance of MAPK pathways to cell proliferation and death is highlighted
by the observation that dysregulation of these kinase cascades can result in
cell transformation and cancer. Activated ERK and JNK pathways can lead to
increased proliferation and survival, although loss of JNK in some instances may
also promote tumorigenesis (Kennedy and Davis, 2003).
In contrast, the p38 MAPK pathway is implicated in suppression of tumorigenesis because it can inhibit cell growth by decreasing the expression of cyclin D (Lavoie et al., 1996), inhibit the activity of Cdc25 phosphatases (Manke et al., 2005), and engage the p16/Rb and p19ARF/p53 tumor suppressor pathways (Bulavin et al., 2002, Bulavin et al., 2004). The p38 MAPK pathway can also cause apoptosis by a mechanism that is incompletely understood but may involve the phosphorylation of members of the Bcl2 family and activation of the mitochondrial apoptotic pathway (Figure 1). The selectivity of the p38 MAPK signaling pathway in tumor suppression is unclear. However, a new study by Dolado et al. (2007) reported in this issue of Cancer Cell now demonstrates that p38 MAPK selectively functions as a sensor of oxidative stress during the initiation of tumorigenesis.
Figure 1Schematic Illustration of the Effects of Reactive Oxygen Species on the p38 MAP Kinase Signal Transduction Pathway
A Radical Role for p38 MAPK in Tumor Initiation: Cancer Cell
https://www.cell.com/cancer-cell/fulltext/S1535-6108(07)00024-4#%20
Biochemistry. 2002 Oct 29;41(43):12995-3002.
Vitamin C suppresses TNF alpha-induced NF kappa B activation
by inhibiting I kappa B alpha phosphorylation.
Cárcamo JM1, Pedraza A, Bórquez-Ojeda O, Golde DW.
Author information
1
Program in Molecular Pharmacology and Chemistry and Department of Clinical
Chemistry, Memorial Sloan-Kettering Cancer Center, 1275 York Avenue, Box 451,
New York, New York 10021, USA.
Abstract
Extracellular stimuli signal for activation of the transcription factor
NFkappaB, leading to gene expression regulating processes involved in immune
responses, inflammation, and cell survival. Tumor necrosis factor-alpha
(TNFalpha) activates NFkappaB via a well-defined kinase pathways involving
NFkappaB-inducing kinase (NIK), which activates downstream multisubunit IkappaB
kinases (IKK). IKK in turn phosphorylates IkappaB, the central regulator of
NFkappaB function.
We found that intracellular vitamin C inhibits TNFalpha-induced activation of NFkappaB in human cell lines (HeLa, monocytic U937, myeloid leukemia HL-60, and breast MCF7) and primary endothelial cells (HUVEC) in a dose-dependent manner. Vitamin C is an important antioxidant, and most cells accumulate ascorbic acid (AA) intracellularly by transporting the oxidized form of the vitamin, dehydroascorbic acid (DHA). Because ascorbic acid is a strong pro-oxidant in the presence of transition metals in vitro, we loaded cells with vitamin C by incubating them with DHA. Vitamin C-loaded cells showed significantly decreased TNFalpha-induced nuclear translocation of NFkappaB, NFkappaB-dependent reporter transcription, and IkappaBalpha phosphorylation.
Our data point to a mechanism of vitamin C suppression of NFkappaB activation by inhibiting TNFalpha-induced activation of NIK and IKKbeta kinases independent of p38 MAP kinase. These results suggest that intracellular vitamin C can influence inflammatory, neoplastic, and apoptotic processes via inhibition of NFkappaB activation.
Vitamin C suppresses TNF alpha-induced NF kappa B activation by inhibiting I
kappa B alpha phosphorylation. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/12390026
蛋白酶和氧化应激控制肺尘上皮细胞中炎性基因表达的有机粉尘诱导
抽象
背景
对有机粉尘中传染源和其他成分的持续炎症反应是肺部损伤和呼吸系统疾病的基础。引起炎症的有机粉尘成分及其引起肺部炎症的机制尚未完全了解。我们研究了家禽粉尘提取物中的蛋白酶活性和细胞内氧化应激诱导A549和Beas2B肺上皮细胞中炎性基因表达的机制。
方法
粉尘提取物对炎症基因表达的影响通过定量聚合酶链反应(qPCR),酶联免疫吸附剂(ELISA)和Western
blot分析进行了分析。通过二氢乙啶(DHE)标记探测氧化应激,并对4-羟基壬烯醛(4-HNE)进行免疫染色。通过瞬时转染测定确定对白介素8(IL-8)启动子调节的影响。
结果
粉尘提取物包含胰蛋白酶和弹性蛋白酶活性,以及活化的蛋白酶活化受体(PAR)-1和-2。丝氨酸蛋白酶抑制剂和PAR-1或PAR-2敲低抑制了炎症基因的诱导。
IL-8基因表达的粉尘提取物诱导与DHE荧光增强和4-HNE染色有关,抗氧化剂抑制了炎症基因的诱导。蛋白酶抑制剂和抗氧化剂可抑制接触粉尘提取物的细胞中的蛋白激酶C和NF-κB活化以及IL-8启动子活性的诱导。
结论
我们的研究表明蛋白酶和细胞内氧化剂控制肺上皮细胞中炎症基因表达的有机粉尘诱导。靶向蛋白酶和氧化应激可能作为治疗有机粉尘诱发的肺部疾病的新方法。这是关于氧化应激参与有机粉尘诱导炎症基因表达的首次报道。
Proteases and oxidant stress control organic dust induction of inflammatory gene
expression in lung epithelial cells
Abstract
Background
Persistant inflammatory responses to infectious agents and other components in
organic dust underlie lung injury and development of respiratory diseases.
Organic dust components responsible for eliciting inflammation and the
mechanisms by which they cause lung inflammation are not fully understood. We
studied the mechanisms by which protease activities in poultry dust extracts and
intracellular oxidant stress induce inflammatory gene expression in A549 and
Beas2B lung epithelial cells.
Methods
The effects of dust extracts on inflammatory gene expression were analyzed by
quantitative polymerase chain reaction (qPCR), enzyme linked immunosorbent
(ELISA) and western blot assays. Oxidant stress was probed by dihydroethidium
(DHE) labeling, and immunostaining for 4-hydroxynonenal (4-HNE). Effects on
interleukin-8 (IL-8) promoter regulation were determined by transient
transfection assay.
Results
Dust extracts contained trypsin and elastase activities, and activated protease
activated receptor (PAR)-1 and -2. Serine protease inhibitors and PAR-1 or PAR-2
knockdown suppressed inflammatory gene induction. Dust extract induction of IL-8
gene expression was associated with increased DHE-fluorescence and 4-HNE
staining, and antioxidants suppressed inflammatory gene induction. Protease
inhibitors and antioxidants suppressed protein kinase C and NF-κB activation and
induction of IL-8 promoter activity in cells exposed to dust extract.
Conclusions
Our studies demonstrate that proteases and intracellular oxidants control
organic dust induction of inflammatory gene expression in lung epithelial cells.
Targeting proteases and oxidant stress may serve as novel approaches for the
treatment of organic dust induced lung diseases. This is the first report on the
involvement of oxidant stress in the induction of inflammatory gene expression
by organic dust.
Proteases and oxidant stress control organic dust induction of inflammatory
gene expression in lung epithelial cells | Respiratory Research | Full Text
https://respiratory-research.biomedcentral.com/articles/10.1186/s12931-016-0455-z
Role of p38 and JNK MAPK signaling pathways and tumor
suppressor p53 on induction of apoptosis in response to Ad-eIF5A1 in A549 lung
cancer cells
1Department of Biology, University of Waterloo, 200 University Ave. W.,
Waterloo, ON N2L 3G1, Canada
Background
The eukaryotic translation initiation factor 5A1 (eIF5A1) is a highly conserved
protein involved in many cellular processes including cell division,
translation, apoptosis, and inflammation. Induction of apoptosis is the only
function of eIF5A1 that is known to be independent of post-translational
hypusine modification. In the present study, we investigated the involvement of
mitogen- and stress-activated protein kinases during apoptosis of A549 lung
cancer cells infected with adenovirus expressing eIF5A1 or a mutant of eIF5A1
that cannot be hypusinated (eIF5A1K50A).
Methods
Using adenoviral-mediated transfection of human A549 lung cancer cells to
over-express eIF5A1 and eIF5A1K50A, the mechanism by which unhypusinated eIF5A1
induces apoptosis was investigated by Western blotting, flow cytometry, and use
of MAPK and p53 inhibitors.
Results
Phosphorylation of ERK, p38 MAPK, and JNK was observed in response to
adenovirus-mediated over-expression of eIF5A1 or eIF5A1K50A, along with
phosphorylation and stabilization of the p53 tumor suppressor protein. Synthetic
inhibitors of p38 and JNK kinase activity, but not inhibitors of ERK1/2 or p53
activity, significantly inhibited apoptosis induced by Ad-eIF5A1. Importantly,
normal lung cells were more resistant to apoptosis induced by eIF5A1 and
eIF5A1K50A than A549 lung cancer cells.
Conclusions
Collectively these data indicate that p38 and JNK MAP kinase signaling are
important for eIF5A1-induced cell death and that induction of apoptosis was not
dependent on p53 activity.
Keywords: eIF5A, Apoptosis, MAPK, p53, Hypusine
MAPKs are serine/threonine protein kinases that participate in intracellular signaling during proliferation, differentiation, cellular stress responses, and apoptosis [18]. Activation of MAPKs, including extracelluar signal-regulated kinases 1 and 2 (ERK1/2), p38 MAPK, and the stress activated protein kinase (SAPK)/c-Jun NH2-terminal kinase (JNK), has been implicated in the activity of numerous chemotherapy and genotoxic drugs. MAPK can regulate apoptosis through specific phosphorylation of downstream mediators of apoptosis, including the tumor suppressor p53, thus linking cellular stress signaling and regulation of p53 activity. Phosphorylation of p53 can regulate p53 activity by altering protein stability, interaction with co-activators, and transcription of target genes [19] as part of the cellular response to stress.
Ad-eIF5A1 and Ad-eIF5AK50A induce activation of ERK kinase, p38 MAPK, and
JNK
Previous studies have demonstrated that treatment with adenovirus eIF5A1 induces
apoptosis in A549 lung carcinoma cells and improves duration of survival in mice
bearing A549 xenograft tumors [14].
Inhibitors of p38 MAPK and JNK protect A549 cells from Ad-eIF5A1-induced
apoptosis
ERK, p38, and JNK signaling pathways are involved in both apoptosis and cell
growth, depending on the cell type and stimulus. The dependence of eIF5A1 on
activation of p38, JNK and ERK for induction of apoptosis was evaluated by
pre-treating A549 cells with specific inhibitors to these kinases and then
inducing apoptosis by infecting the cells with Ad-eIF5A1 (Figure 6). Since
Ad-eIF5A1 infection is associated with increased expression and activity of p53
(Figures 3, ,4,4, and and5),5), cells were also pre-treated with pifithrin-α
in order to determine whether eIF5A1-induced apoptosis is dependent on p53
activity in A549 cells. MEK inhibition did not significantly affect induction of
apoptosis by Ad-eIF5A1. Inhibition of p38 and JNK both significantly reduced
eIF5A1-induced apoptosis while use of both inhibitors in combination inhibited
apoptosis by approximately 50% (p < 0.001), suggesting that activation of p38
and JNK are both important in the induction of apoptosis by eIF5A1 (Figure 6).
Inhibition of p53 activity did not impact apoptosis resulting from Ad-eIF5A1
infection suggesting that, although p53 is up-regulated in response to eIF5A1,
it is not required for apoptosis (Figure 6).
Normal lung fibroblasts are resistant to Ad-eIF5A1-induced apoptosis
The ability to kill malignant cells without harming normal cells is an important
feature of an ideal cancer therapy drug.
Conclusions
In summary, this study has identified the activation of MAPKs as an important
step in the signaling cascade that leads to the induction of p53-independent
apoptotic cell death in response to over-expression of unhypusinated eIF5A1 in
A549 lung carcinoma cells. The importance of p38 and JNK activation during
eIF5A1-induced apoptosis is highlighted by the ability of inhibitors of these
MAPKs to inhibit apoptosis ensuing from Ad-eIF5A1 infection. Furthermore,
malignant A549 cells demonstrated enhanced sensitivity to eIF5A1-induced
apoptosis compared to normal lung cells, suggesting that eIF5A1-based therapy
may spare normal tissues. This work emphasizes the potential of therapeutic
application of eIF5A1 in the treatment in cancers.
Role of p38 and JNK MAPK signaling pathways and tumor suppressor p53 on
induction of apoptosis in response to Ad-eIF5A1 in A549 lung cancer cells
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3660295/
新合成的2-(3-羟基-5-甲氧基苯基)-6,7-亚甲基二氧基喹啉-4-酮通过诱导HL-60人白血病细胞的氧化应激和上调p38
MAPK信号通路来触发细胞凋亡。
The newly synthesized
2-(3-hydroxy-5-methoxyphenyl)-6,7-methylenedioxyquinolin-4-one triggers cell
apoptosis through induction of oxidative stress and upregulation of the p38 MAPK
signaling pathway in HL-60 human leukemia cells
摘要
本研究的目的是发现与2-(3-羟基-5-甲氧基苯基)-6,7-亚甲基二氧基喹啉-4-酮(YYK1)诱导的HL-60人白血病细胞凋亡相关的信号通路。
YYK1诱导HL-60细胞的细胞毒性作用,细胞形态变化,减少细胞数量,增加活性氧(ROS)的产生以及线粒体膜电位(ΔΨm)的损失。末端脱氧核苷酸转移酶dUTP缺口末端标记(TUNEL)染色证实了YYK1诱导的细胞凋亡。比色分析和蛋白质印迹分析的结果表明,在YYK1处理的HL-60细胞中,caspase-7
/ -3,caspase-8和caspase-9的活性增加。蛋白质印迹分析表明,外在凋亡蛋白(Fas /
CD95,FasL和FADD),内在相关蛋白(细胞色素c,Apaf-1,AIF和Endo G)的蛋白水平,Bax /
Bcl-2和磷酸化p38的比率YYK1处理后,HL-60细胞中的MAPK增加。用N-乙酰基半胱氨酸(NAC; ROS清除剂)或二苯撑碘鎓氯化物(DPI;
NADPH氧化酶抑制剂)预处理后,细胞凋亡显着减少。 SB202190对p38 MAPK信号的阻断消除了YYK1诱导的HL-60细胞Fas /
CD95上调和细胞凋亡。我们得出的结论是,YYK1通过ROS介导的HL-60人白血病细胞中p38 MAPK信号转导的激活,诱导了外在和内在的凋亡途径。
细胞凋亡的特征是形态变化,细胞收缩和染色质凝集(1-3)。凋亡涉及两个信号途径。内在途径包括破坏线粒体膜,然后将细胞色素c,凋亡蛋白酶激活因子-1(Apaf-1),半胱天冬酶原9,凋亡诱导因子(AIF)和核酸内切酶G(Endo
G)释放到细胞质中。通过死亡受体和配体相互作用(例如FasL /
Fas)然后激活caspase-8的外部途径(4,5)。内在和外在途径均诱导了caspase-7和caspase-3的活化(6)。最近的一项研究还表明,促分裂原激活的蛋白激酶(MAPKs)信号传导能够调节肿瘤细胞中与凋亡相关的通路(7,8)。
MAPKs信号调节生理功能,包括细胞增殖,分化,发育和凋亡(9,10)。 MAPKs有三个主要的亚家族,包括细胞外信号调节激酶(ERK),c-Jun
N端激酶(JNK)和p38蛋白(11)。诸如生长因子,激素或细胞因子之类的刺激会激活MAP激酶激酶(MKK)。
MKK将MAPK的酪氨酸/苏氨酸残基磷酸化,然后导致MAPK的二聚化和随后的活化(12)。
ERK级联最常与细胞存活活性有关,但p38和JNK蛋白似乎具有促凋亡作用(11,13)。 p38
MAPK被多种细胞应激激活,并在线粒体功能异常和胱天蛋白酶激活细胞凋亡之前的较早阶段起作用(14)。最近的研究表明,p38
MAPK激酶级联在细胞应激引起的细胞凋亡过程中是必需的(14,15)。
在本研究中,我们设计并合成了一种新型的抗白血病化合物2-(3-羟基-5-甲氧基苯基)-6,7-亚甲基二氧基喹啉-4-酮(YYK1),它是抗白血病最有潜力的候选药物白血病活性如图1所示。但是,尚未充分研究YYK1对HL-60白血病细胞的细胞毒作用或抗白血病活性的分子机制。在这项研究中,增殖曲线表明YYK1对HL-60细胞的细胞毒性具有时间和浓度依赖性。
YYK1导致凋亡细胞死亡,随后是ROS的产生,p38 MAPK的持续活化。我们的研究结果强烈表明p38
MAPK通路在YYK1诱导的HL-60细胞凋亡引起的细胞死亡过程中具有必要的作用。
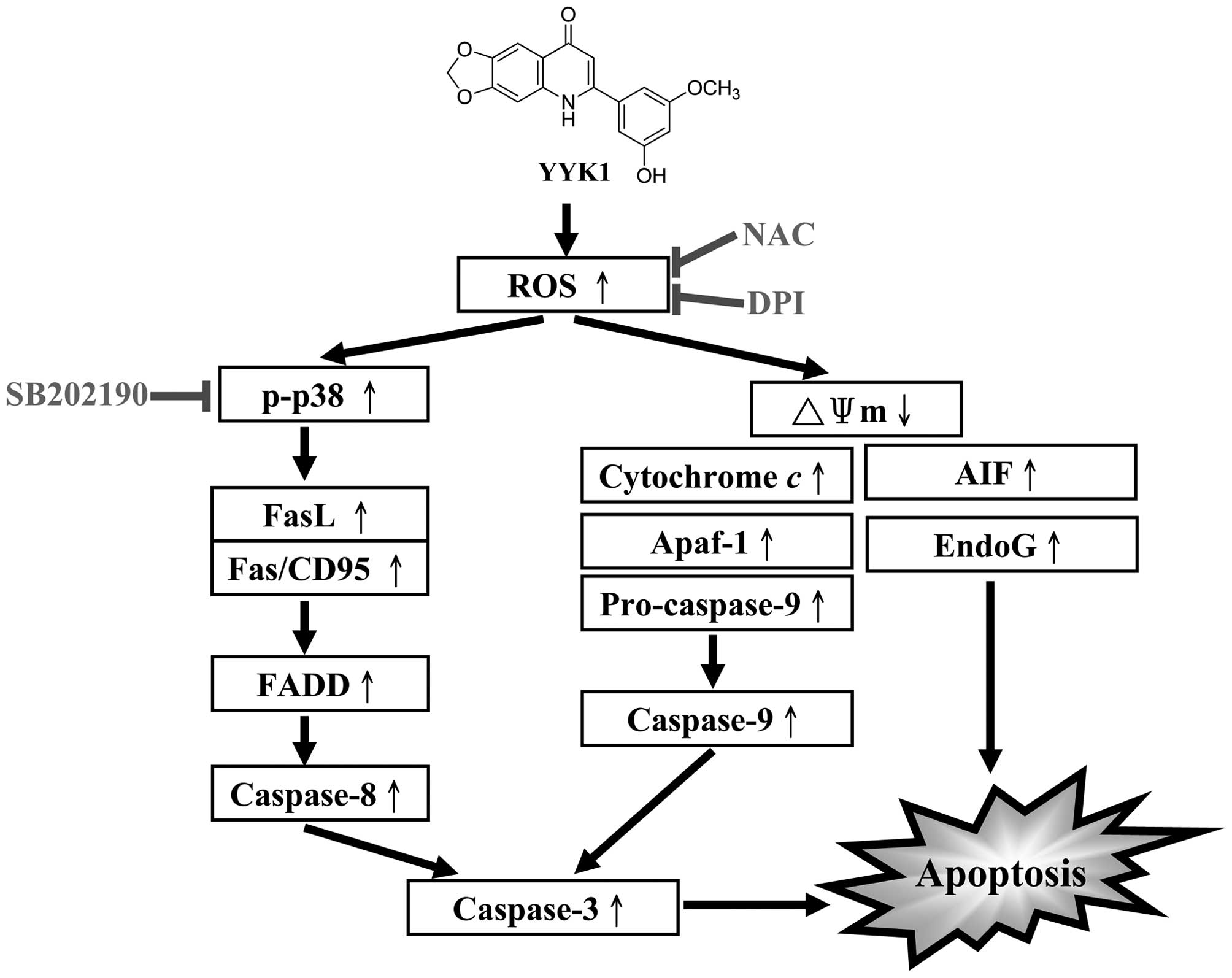
The newly synthesized
2-(3-hydroxy-5-methoxyphenyl)-6,7-methylenedioxyquinolin-4-one triggers cell
apoptosis through induction of oxidative stress and upregulation of the p38 MAPK
signaling pathway in HL-60 human leukemia cells
Abstract
The aim of the present study was to discover the signaling pathways associated
with 2-(3-hydroxy-5-methoxyphenyl)-6,7-methylenedioxyquinolin-4-one
(YYK1)-induced apoptosis in HL-60 human leukemia cells. YYK1 induced cytotoxic
effects, cell morphological changes, decreased the cell number and increased
reactive oxygen species (ROS) production and loss of mitochondrial membrane
potential (ΔΨm) in HL-60 cells. YYK1-induced apoptosis was confirmed by the
terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining.
Results from colorimetric assays and western blot analysis indicated that
activities of caspase-7/-3, caspase-8 and caspase-9 were increased in
YYK1-treated HL-60 cells. Western blot analysis showed that the protein levels
of extrinsic apoptotic proteins (Fas/CD95, FasL and FADD), intrinsic related
proteins (cytochrome c, Apaf-1, AIF and Endo G), the ratio of Bax/Bcl-2 and
phosphorylated p38 MAPK were increased in HL-60 cells after YYK1 treatment. Cell
apoptosis was significantly reduced after pre-treatment with N-acetylcysteine
(NAC; a ROS scavenger) or diphenyleneiodonium chloride (DPI; a NADPH oxidase
inhibitor). Blockage of p38 MAPK signaling by SB202190 abolished YYK1-induced
Fas/CD95 upregulation and apoptosis in HL-60 cells. We conclude that YYK1
induces both of extrinsic and intrinsic apoptotic pathways via ROS-mediated
activation of p38 MAPK signaling in HL-60 human leukemia cells in vitro.
Apoptosis is characterized by morphological changes, cell shrinkage and
chromatin condensation (1–3). Two signal pathways are involved in apoptosis. The
intrinsic pathway involves disrupting the mitochondrial membrane and then
releasing cytochrome c, apoptotic protease activating factor-1 (Apaf-1),
pro-caspase-9, apoptosis-inducing factor (AIF) and endonuclease G (Endo G) into
the cytosol. The extrinsic pathway through death receptors and ligand
interaction such as FasL/Fas and then activate caspase-8 (4,5). Both of
intrinsic and extrinsic pathways induced the activation of caspase-7 and
caspase-3 (6). A recent study has also demonstrated that the mitogen-activated
protein kinases (MAPKs) signaling is able to regulate apoptosis-associated
pathways in tumor cells (7,8).
The MAPKs signaling modulate physiological functions and include cell
proliferation, differentiation, development and apoptosis (9,10). There are
three major subfamilies of MAPKs including extracellular signal-regulated kinase
(ERK), c-Jun N-terminal kinase (JNK) and p38 proteins (11). Stimulations such as
growth factors, hormones, or cytokines activate MAP kinase kinases (MKKs). MKKs
phosphorylate tyrosine/threonine residues of MAPKs then result in dimerization
and subsequent activation of MAPKs (12). The ERK cascade is most frequently
associated with cell survival activity but the p38 and JNK proteins appear to be
pro-apoptotic effects (11,13). p38 MAPK is activated by a variety of cellular
stresses and acts at early step prior to dysfunction of mitochondria and caspase
activation in cell apoptosis (14). The recent studies have demonstrated that the
p38 MAPK kinase cascade is necessary during cell apoptosis by cellular stress
(14,15).
In the present study, we designed and synthesized a novel anti-leukemia
compound, 2-(3-hydroxy-5-methoxyphenyl)-6,7-methylenedioxyquinolin-4-one (YYK1),
it is a most potential candidate for anti-leukemia activities as shown in Fig.
1. However, neither the cytotoxic effects of YYK1 on HL-60 leukemia cells nor
the molecular mechanisms underlying its anti-leukemia activity have been well
investigated. In this study, the proliferation curve showed that YYK1 had time-
and concentration-dependent effects on the cytotoxicity of HL-60 cells. YYK1
caused apoptotic cell death, which is preceded by the generation of ROS,
sustained activation of p38 MAPK. Our results strongly suggest a necessary role
for the p38 MAPK pathway during cell death by apoptosis in HL-60 cells induced
by YYK1.
https://www.spandidos-publications.com/10.3892/or.2012.1923
黄连素通过p38激活KB人口腔癌细胞诱导FasL相关凋亡
摘要
在本研究中,我们检查了黄连素碱在KB口腔癌细胞中的抗癌特性,特别关注其细胞机制。黄连素不影响用作对照的人类原代正常口腔角质形成细胞的细胞活力。然而,发现在黄连素存在下,KB细胞的活力以剂量依赖性方式显着降低。此外,在KB细胞中,黄连素诱导了基因组DNA的断裂,细胞形态的变化和核浓缩。另外,观察到caspase-3和-7活化,并且凋亡增加。黄连素碱还被发现显着上调死亡受体配体FasL的表达。反过来,这种上调触发了促凋亡因子如caspase-8,-9和-3以及聚(ADP-核糖)聚合酶(PARP)的激活。此外,黄连素还显着上调了促凋亡因子,例如Bax,Bad和Apaf-1。
Bcl-2和Bcl-xL等抗凋亡因子被下调。
Z-VAD-FMK是一种可透过细胞的泛半胱天冬酶抑制剂,可抑制caspase-3和PARP的活化。这些结果清楚地表明,黄连素诱导的KB口腔癌细胞死亡是由外源性死亡受体依赖性和内在的线粒体依赖性凋亡信号通路介导的。此外,黄连素诱导的FasL上调被证明是由p38
MAPK信号通路介导的。我们还发现,黄连素诱导的迁移抑制是通过p38
MAPK磷酸化下调MMP-2和MMP-9介导的。总而言之,黄连素有潜力被用作治疗口腔癌的具有有限副作用的化学治疗剂。
Berberine induces FasL-related apoptosis through p38
activation in KB human oral cancer cells
Abstract
In the present study, we examined the anticancer properties of berberine in KB
oral cancer cells with a specific focus on its cellular mechanism. Berberine did
not affect the cell viability of the primary human normal oral keratinocytes
that were used as a control. However, the viability of KB cells was found to
decrease significantly in the presence of berberine in a dose-dependent manner.
Furthermore, in KB cells, berberine induced the fragmentation of genomic DNA,
changes in cell morphology, and nuclear condensation. In addition, caspase-3 and
-7 activation, and an increase in apoptosis were observed. Berberine was also
found to upregulate significantly the expression of the death receptor ligand,
FasL. In turn, this upregulation triggered the activation of pro-apoptotic
factors such as caspase-8, -9 and -3 and poly(ADP-ribose) polymerase (PARP).
Furthermore, pro-apoptotic factors such as Bax, Bad and Apaf-1 were also
significantly upregulated by berberine. Anti-apoptotic factors such as Bcl-2 and
Bcl-xL were downregulated. Z-VAD-FMK, a cell-permeable pan-caspase inhibitor,
suppressed the activation of caspase-3 and PARP. These results clearly indicate
that berberine-induced cell death of KB oral cancer cells was mediated by both
extrinsic death receptor-dependent and intrinsic mitochondrial-dependent
apoptotic signaling pathways. In addition, berberine-induced upregulation of
FasL was shown to be mediated by the p38 MAPK signaling pathway. We also found
that berberine-induced migration suppression was mediated by downregulation of
MMP-2 and MMP-9 through phosphorylation of p38 MAPK. In summary, berberine has
the potential to be used as a chemotherapeutic agent, with limited side-effects,
for the management of oral cancer.
Keywords: berberine, oral cancer, apoptosis, migration, chemotherapy
Oncol Rep. 2015 Apr; 33(4): 1775–1782.
1The Division of Natural Medical Science, College of Health Science, Chosun
University, Dong-gu, Gwangju 501-759, Republic of Korea
2Oral Biology Research Institute, Chosun University, Dong-gu, Gwangju 501-759,
Republic of Korea
3Regional Innovation Center for Dental Science and Engineering, Chosun
University, Dong-gu, Gwangju 501-759, Republic of Korea
4Department of Oral and Maxillofacial Surgery, School of Dentistry, Chosun
University, Dong-gu, Gwangju 501-759, Republic of Korea
5Department of Prosthodontics, School of Dentistry, Chosun University, Dong-gu,
Gwangju 501-759, Republic of Korea
6Department of Biochemistry, Rush University Medical Center, Chicago, IL 60612,
USA
Correspondence to: Professor Su-Gwan Kim, Department of Oral and Maxillofacial
Surgery, School of Dentistry, Chosun University, 375 Seosuk-dong, Dong-gu,
Gwangju 501-759, Republic of Korea, E-mail: rk.ca.nusohc@mikcgs
Berberine induces FasL-related apoptosis through p38 activation in KB human oral
cancer cells
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4440222/
Regulation of FAS Ligand Expression during
Activation-Induced Cell Death in T Cells by p38 Mitogen-Activated Protein Kinase
and C-Jun Nh2-Terminal Kinase
Abstract
Activation-induced cell death (AICD) is a mechanism of peripheral T cell
tolerance that depends upon an interaction between Fas and Fas ligand (FasL).
Although c-Jun NH2-terminal kinase (JNK) and p38 mitogen-activated protein
kinase (MAPK) may be involved in apoptosis in various cell types, the mode of
regulation of FasL expression during AICD in T cells by these two MAPKs is
incompletely understood. To investigate the regulatory roles of these two MAPKs,
we analyzed the kinetics of TCR-induced p38 MAPK and JNK activity and their
regulation of FasL expression and AICD. We report that both JNK and p38 MAPK
regulate AICD in T cells. Our data suggest a novel model of T cell AICD in which
p38 MAPK acts early to initiate FasL expression and the Fas-mediated activation
of caspases. Subsequently, caspases stimulate JNK to further upregulate FasL
expression. Thus, p38 MAPK and downstream JNK converge to regulate FasL
expression at different times after T cell receptor stimulation to elicit
maximum AICD.
Keywords: Fas ligand, apoptosis, p38 MAPK, JNK, T cells
Regulation of FAS Ligand Expression during Activation-Induced Cell Death in T
Cells by p38 Mitogen-Activated Protein Kinase and C-Jun Nh2-Terminal Kinase
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2193110/
Curcumin induces FasL-related apoptosis through p38 activation in human
hepatocellular carcinoma Huh7 cells
Aim
The aim of this study is to explore the underlying molecular mechanism of
curcumin-induced apoptosis in human hepatocellular carcinoma (HCC) Huh7 cells.
Main methods
Fas and FasL mRNA expression was analyzed by reverse transcription PCR. Western
blot was applied to detect the protein expression of Bcl-2 family members, MAPK
family members, c-Jun, c-Fos, ATF-2, caspase-3, PARP, TNF receptor family
members and the respective ligands. Apoptotic cells were assayed with annexin
V/PI double staining and flow cytometry.
Key findings
Curcumin treatment resulted in a fast and significant increase of Fas and Fas
ligand (FasL) along with activation of caspase-3 and cleavage of PARP in Huh7
cells. Inhibition of caspase-3 activity by the specific inhibitor Z-DEVD-FMK
rescued Huh7 cells from curcumin-induced apoptosis. Neutralization of FasL
significantly protected the cells from curcumin-induced caspase-3 activation and
apoptosis in a dose-dependent manner. Moreover, p38 was rapidly activated in
response to curcumin, and inactivation of p38 by pharmacologic inhibitor
SB203580 dramatically suppressed curcumin-induced FasL expression and apoptosis.
Significance
Our results demonstrated that curcumin induces apoptosis through p38-denpendent
up-regulation of FasL in Huh7 cells.
Curcumin induces FasL-related apoptosis through p38 activation in human
hepatocellular carcinoma Huh7 cells - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S0024320513000234
Arachidonic acid induces Fas and FasL upregulation in human
leukemia U937 cells via Ca2+/ROS-mediated suppression of ERK/c-Fos pathway and
activation of p38 MAPK/ATF-2 pathway
Arachidonic acid (AA)-induced apoptotic death of human leukemia U937 cells was
characteristic of increase in intracellular Ca2+ concentration ([Ca2+]i), ROS
generation, ERK inactivation, p38 MPAK activation, degradation of procaspase-8
and production of truncated Bid (tBid). Moreover, AA treatment upregulated
Fas/FasL protein expression and transcription of Fas/FasL mRNA. Downregulation
of FADD blocked AA-induced procaspase-8 degradation and rescued viability of
AA-treated cells. BAPTA-AM (Ca2+ chelator) pretreatment abolished AA-induced ROS
generation, while N-acetylcystein (NAC, ROS scavenger) was unable to alter
AA-elicited [Ca2+]i increase. Pretreatment with BAPTA-AM or NAC abrogated p38
MAPK activation and restored ERK activation. Suppression of p38 MAPK or
transfection of constitutively active MEK1 abolished AA-induced Fas and FasL
upregulation. AA treatment repressed ERK-mediated c-Fos phosphorylation but
evoked p38 MAPK-mediated ATF-2 phosphorylation. Knockdown of c-Fos and ATF-2 by
siRNA reflected that c-Fos counteracted the effect of ATF-2 on Fas/FasL
upregulation. Taken together, our data indicate that Fas/FasL upregulation in
AA-treated U937 cells is elicited by Ca2+/ROS-mediated suppression of ERK/c-Fos
pathway and activation of p38 MAPK/ATF-2, and suggest that autocrine
Fas-mediated apoptotoic mechanism is involved in AA-induced cell death.
Arachidonic acid induces Fas and FasL upregulation in human leukemia U937
cells via Ca2+/ROS-mediated suppression of ERK/c-Fos pathway and activation of
p38 MAPK/ATF-2 pathway - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S0378427409014015
Working model of TNF- ␣ - and FAS ligand–induced neutrophil apoptosis
Working model of TNF- ␣ - and FAS ligand–induced neutrophil apoptosis. Left panel shows that TNFR1 ligation results in p38 activation. p38 further activates the PI3K signaling pathway and ROS generation. ROS increase caspase-3 activity, leading to apoptosis. Right panel shows that ligation of FAS results in caspase-8 processing, which further activates caspase-3 and BID. BID cleavage launches the mitochondrial amplification loop, leading to mitochondrial outer membrane permeabilization, cytochrome c release, apoptosome formation, and caspase-3 activation, resulting in apoptosis.
Working model of TNF- ␣ - and FAS ligand–induced neutrophil apoptosis.... |
Download Scientific Diagram
https://www.researchgate.net/figure/Working-model-of-TNF--and-FAS-ligand-induced-neutrophil-apoptosis-Left-panel-shows_fig6_51037453
... contrast, TNF-stimulation of neutrophils did not result in detectable caspase-8 cleavage and activation, whereas it did induce caspase-3 activity ( Figure 6A-B). TNF--induced caspase-3 pro- cessing was not blocked by inhibitors of p38 and PI3Ks ( Figure 6A); however, blocking of p38 or PI3Ks resulted in reduced caspase-3 activity ( Figure 6B) and lamin B cleavage (supplemental Figure 7). These data suggest that ROS are required for full caspase-3 activation. ...
.. ROS activate either directly or indirectly effector caspases. The proximal
events leading to activation of the caspase cascade differ largely between TNFR1
and FAS stimulation within neutro- phils ( Figure 7). Moreover, our data reveal
major differences in TNFR1 signaling in neutrophils compared with published
results from cells with less active NADPH oxidase. ...
View
Role of P38 Mitogen-Activated Protein Kinase Phosphorylation and Fas-Fas Ligand Interaction in Morphine-Induced Macrophage Apoptosis | The Journal of Immunology
Previous reports suggest that morphine-induced macrophage apoptosis is
mediated through opiate receptors (14). This study delineates the involved
mechanisms. Naltrexone, an opiate receptor antagonist, inhibited the effect of
morphine on P38 MAPK phosphorylation and FasL expression. Because both FasL
expression and P38 MAPK phosphorylation in response to morphine are key events
in the induction of macrophage apoptosis it appears that opiate receptors play a
role in morphine-induced macrophage apoptosis.
We conclude that morphine promotes P38 MAPK phosphorylation via opiate receptors
through TGF-β- and iNOS-mediated downstream signaling. Because morphine
activates proteins involved in extrinsic as well as intrinsic cell death
pathways it appears that both pathways contribute to morphine-induced macrophage
apoptosis.
https://www.jimmunol.org/content/168/8/4025.full

Frontiers | The potential of Fas ligand (apoptosis-inducing molecule) as an
unconventional therapeutic target in type 1 diabetes | Immunology
https://www.frontiersin.org/articles/10.3389/fimmu.2012.00196/full
1 Department of Pathology, Johns Hopkins University School of Medicine,
Baltimore, MD, USA
2 Department of Pediatrics, Johns Hopkins University School of Medicine,
Baltimore, MD, USA
3 Department of Medicine, Johns Hopkins University School of Medicine,
Baltimore, MD, US
Melanoma Cell Expression of Fas(Apo-1/CD95) Ligand ...
https://science.sciencemag.org/content/274/5291/1363
Nov 22, 1996 · Malignant melanoma accounts for most of the increasing
mortality from skin cancer. Melanoma cells were found to express Fas (also
called Apo-1 or CD95) ligand (FasL). In metastatic lesions, Fas-expressing T
cell infiltrates were proximal to FasL+ tumor cells. In vitro, apoptosis of
Fas-sensitive target cells occurred upon incubation with melanoma tumor cells;
and in vivo, injection of … Cited by: 1599 Publish Year: 1996 Author: M. Hahne,
D. Rimoldi, M. Schröter, P. Romero, M. Sc
Expression of FASL in the Tumor Endothelium Limits T-cell Infiltration
Ovarian Cancer Research Center, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania, USA.
DOI: 10.1158/2159-8290.CD-RW2014-106 Published July 2014
ArticleInfo & Metrics
Major finding: Fas ligand (FASL)-expressing tumor vasculature restricts CD8+
T-cell infiltration into tumors.
Mechanism: Tumor-derived VEGF, IL10, and PGE2 induce endothelial FASL expression
to kill effector T cells.
Impact: Reduction of endothelial FASL through VEGF and PGE2 inhibition may
restore antitumor immune responses.
The tumor endothelium integrates angiogenic and immunosuppressive signals to
limit infiltration of T cells into tumors and promote immune evasion.
Given that FAS ligand (FASL) is a known mediator of T- cell apoptosis that has been shown to be expressed on the tumor endothelium, Motz and colleagues evaluated the role of FASL in regulation of the tumor endothelial barrier. FASL was highly expressed in tumor blood vessels across six types of cancers compared with normal vasculature and most tumor cells. FASL-expressing endothelial cells were capable of killing effector T cells, but regulatory T cells were resistant to FASL, suggesting that FASL suppresses antitumor immune responses by shifting the balance between effector T cells and Tregs in tumors. Consistent with this finding, endothelial FASL expression was associated with a lower number of infiltrating CD8+ T cells across several human solid tumor types. The authors screened tumor-derived soluble factors for the ability to upregulate FASL in endothelial cells and found that IL10, prostaglandin E2 (PGE2), and VEGF cooperated to induce FASL expression in human microvascular endothelia. Combined pharmacologic blockade of PGE2 production with acetylsalicylic acid (aspirin) and VEGFA inhibition with a VEGF-neutralizing antibody effectively abrogated endothelial FASL induction by ovarian cancer cell supernatants and suppressed both FASL expression on tumor endothelial cells and tumor growth in multiple xenograft mouse models in a CD8+ T-cell–dependent manner. Additionally, blockade of endothelial FASL signaling enhanced the efficacy of adoptive transfer of antitumor T cells from syngeneic mice and prolonged survival. Therefore, in addition to highlighting a central role of endothelial FASL expression in tumor immune tolerance, these findings suggest that combined strategies that target angiogenic and immunosuppressive mechanisms will be needed to effectively inhibit tumor progression.
Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med 2014 May 4 [Epub ahead of print].
Tumor endothelium FasL establishes a selective immune barrier promoting
tolerance in tumors
Gregory T Motz, Stephen P Santoro, Li-Ping Wang, Tom Garrabrant, Ricardo R
Lastra, Ian S Hagemann, Priti Lal, Michael D Feldman, Fabian Benencia & George
Coukos
Nature Medicine volume 20, pages 607–615 (2014) | Download Citation
Abstract
We describe a new mechanism regulating the tumor endothelial barrier and T cell
infiltration into tumors. We detected selective expression of the death mediator
Fas ligand (FasL, also called CD95L) in the vasculature of human and mouse solid
tumors but not in normal vasculature. In these tumors, FasL expression was
associated with scarce CD8+ infiltration and a predominance of FoxP3+ T
regulatory (Treg) cells. Tumor-derived vascular endothelial growth factor A
(VEGF-A), interleukin 10 (IL-10) and prostaglandin E2 (PGE2) cooperatively
induced FasL expression in endothelial cells, which acquired the ability to kill
effector CD8+ T cells but not Treg cells because of higher levels of c-FLIP
expression in Treg cells. In mice, genetic or pharmacologic suppression of FasL
produced a substantial increase in the influx of tumor-rejecting CD8+ over
FoxP3+ T cells. Pharmacologic inhibition of VEGF and PGE2 produced a marked
increase in the influx of tumor-rejecting CD8+ over FoxP3+ T cells that was
dependent on attenuation of FasL expression and led to CD8-dependent tumor
growth suppression. Thus, tumor paracrine mechanisms establish a tumor
endothelial death barrier, which has a critical role in establishing immune
tolerance and determining the fate of tumors.
https://www.nature.com/articles/nm.3541
Expression of FASL in the Tumor Endothelium Limits T-cell Infiltration | Cancer
Discovery
https://cancerdiscovery.aacrjournals.org/content/4/7/OF13
Lactic Acid Upregulates VEGF Expression in Macrophages and Facilitates
Choroidal Neovascularization.
Song J1,2, Lee K3, Park SW4, Chung H1, Jung D1, Na YR1, Quan H1, Cho CS4, Che
JH5, Kim JH4, Park JH2, Seok SH1.
Author information
1
Department of Microbiology and Immunology, Institute of Endemic Disease, Seoul
National University College of Medicine, Chongno-gu, Seoul, South Korea.
2
Department of Laboratory Animal Medicine, College of Veterinary Medicine, Seoul
National University, Gwanak-gu, Seoul, South Korea.
3
Department of Ophthalmology, Ajou University School of Medicine, Suwon-si, South
Korea.
4
FARB Laboratory, Biomedical Research Institute, Seoul National University
Hospital, Seoul, South Korea.
5
Biomedical Research Institute, Seoul National University Hospital, Chongno-gu,
Seoul, South Korea.
Abstract
PURPOSE:
Lactic acid, the end product of glycolysis, has emerged as an immune-modulating
metabolite in various diseases. In this study, we aimed to examine whether
lactic acid contributes to the disease pathogenesis of choroidal
neovascularization (CNV) and to investigate the role of macrophages in CNV
pathogenesis.
METHODS:
CNV was induced by laser photocoagulation in C57BL/6J mice. Lactic acid
concentration was measured in the RPE-choroid region. Macrophage infiltration
and VEGF were quantified by flow cytometry. VEGF-positive areas and CNV lesions
were measured by flat-mount immunofluorescence staining. To inhibit lactic acid
uptake in vivo, alpha-cyano-4-hydroxycinnamic acid (α-CHC), a monocarboxylate
transporter (MCT) blocker, was injected intravitreally 1 day after laser. VEGF
productions were measured in ARPE-19, THP-1 cells, and human umbilical vein
endothelial cells (HUVECs) by quantitative PCR and ELISA. Angiogenic activity of
lactic acid-treated macrophages was assessed by HUVEC tube formation assay.
RESULTS:
Lactic acid was significantly increased in the RPE-choroid region of CNV-induced
mice. Lactic acid upregulated VEGFA mRNA and VEGF protein expressions in THP-1
macrophages, but did not in ARPE-19 or HUVECs. THP-1 macrophages treated with
lactic acid increased the angiogenesis of endothelial cells independent of MCT
activity. Intravitreal injection of α-CHC substantially reduced the
VEGF-positive area that colocalized with F4/80-positive macrophages. CNV lesions
were also significantly reduced following α-CHC injection compared with
vehicle-injected controls.
CONCLUSIONS:
To our knowledge, these results show for the first time the role of lactic acid
in facilitating neovascularization through macrophage-induced angiogenesis. We
suggest that targeting macrophage metabolism can be a promising strategy for CNV
treatment.
Lactic Acid Upregulates VEGF Expression in Macrophages and Facilitates Choroidal
Neovascularization. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/30046816